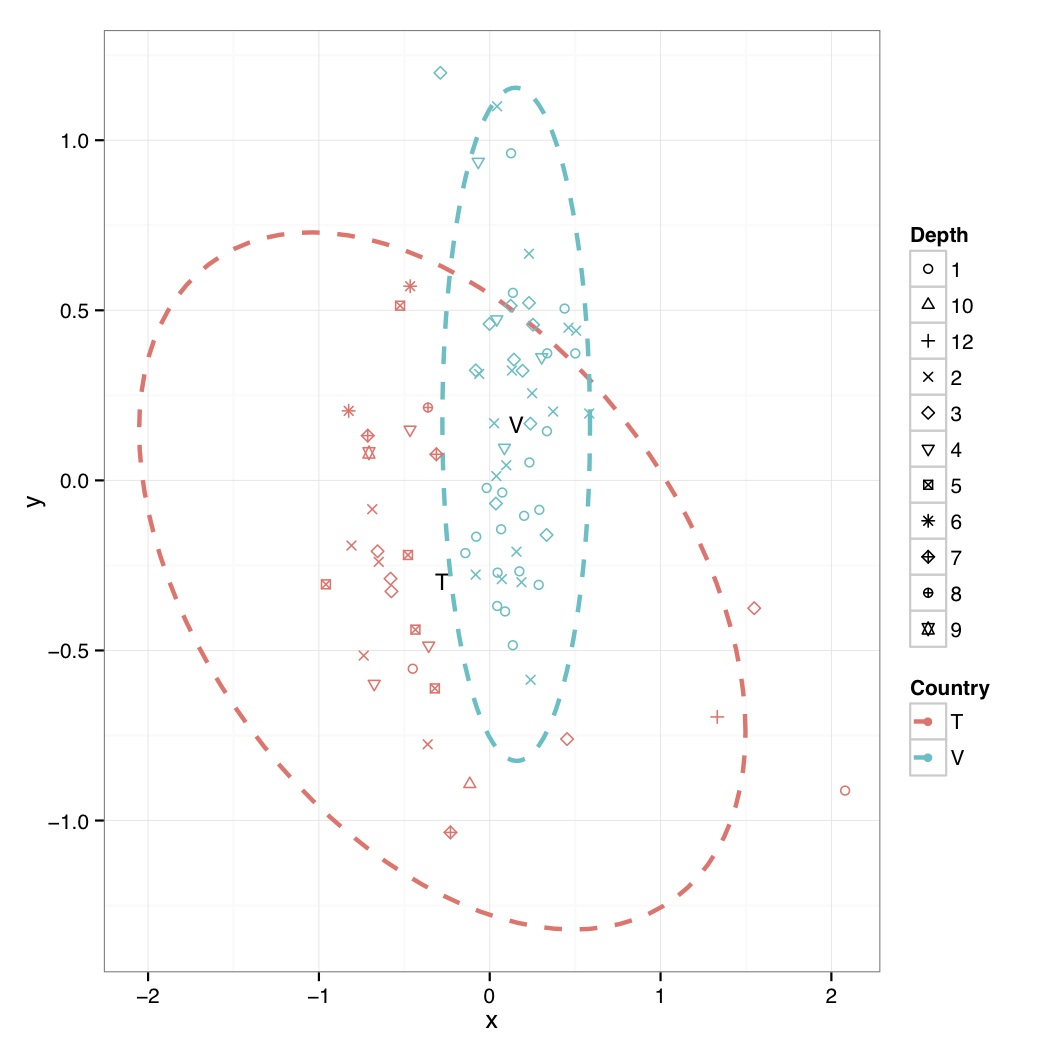
# ============================================================ # Tutorial on drawing an NMDS plot using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) meta_table<-read.csv("ENV_pitlatrine.csv",row.names=1,check.names=FALSE) #Just a check to ensure that the samples in meta_table are in the same order as in abund_table meta_table<-meta_table[rownames(abund_table),] #Get grouping information grouping_info<-data.frame(row.names=rownames(abund_table),t(as.data.frame(strsplit(rownames(abund_table),"_")))) # > head(grouping_info) # X1 X2 X3 # T_2_1 T 2 1 # T_2_10 T 2 10 # T_2_12 T 2 12 # T_2_2 T 2 2 # T_2_3 T 2 3 # T_2_6 T 2 6 #Load vegan library library(vegan) #Get MDS stats sol<-metaMDS(abund_table,distance = "bray", k = 2, trymax = 50) #Make a new data frame, and put country, latrine, and depth information there, to be useful for coloring, and shape of points NMDS=data.frame(x=sol$point[,1],y=sol$point[,2],Country=as.factor(grouping_info[,1]),Latrine=as.factor(grouping_info[,2]),Depth=as.factor(grouping_info[,3])) #Get spread of points based on countries plot.new() ord<-ordiellipse(sol, as.factor(grouping_info[,1]) ,display = "sites", kind ="sd", conf = 0.95, label = T) dev.off() #Reference: http://stackoverflow.com/questions/13794419/plotting-ordiellipse-function-from-vegan-package-onto-nmds-plot-created-in-ggplo #Data frame df_ell contains values to show ellipses. It is calculated with function veganCovEllipse which is hidden in vegan package. This function is applied to each level of NMDS (group) and it uses also function cov.wt to calculate covariance matrix. veganCovEllipse<-function (cov, center = c(0, 0), scale = 1, npoints = 100) { theta <- (0:npoints) * 2 * pi/npoints Circle <- cbind(cos(theta), sin(theta)) t(center + scale * t(Circle %*% chol(cov))) } #Generate ellipse points df_ell <- data.frame() for(g in levels(NMDS$Country)){ if(g!="" && (g %in% names(ord))){ df_ell <- rbind(df_ell, cbind(as.data.frame(with(NMDS[NMDS$Country==g,], veganCovEllipse(ord[[g]]$cov,ord[[g]]$center,ord[[g]]$scale))) ,Country=g)) } } # > head(df_ell) # NMDS1 NMDS2 Country # 1 1.497379 -0.7389216 T # 2 1.493876 -0.6800680 T # 3 1.483383 -0.6196981 T # 4 1.465941 -0.5580502 T # 5 1.441619 -0.4953674 T # 6 1.410512 -0.4318972 T #Generate mean values from NMDS plot grouped on Countries NMDS.mean=aggregate(NMDS[,1:2],list(group=NMDS$Country),mean) # > NMDS.mean # group x y # 1 T -0.2774564 -0.2958445 # 2 V 0.1547353 0.1649902 #Now do the actual plotting library(ggplot2) shape_values<-seq(1,11) p<-ggplot(data=NMDS,aes(x,y,colour=Country)) p<-p+ annotate("text",x=NMDS.mean$x,y=NMDS.mean$y,label=NMDS.mean$group,size=4) p<-p+ geom_path(data=df_ell, aes(x=NMDS1, y=NMDS2), size=1, linetype=2) p<-p+geom_point(aes(shape=Depth))+scale_shape_manual(values=shape_values)+theme_bw() pdf("NMDS.pdf") print(p) dev.off()
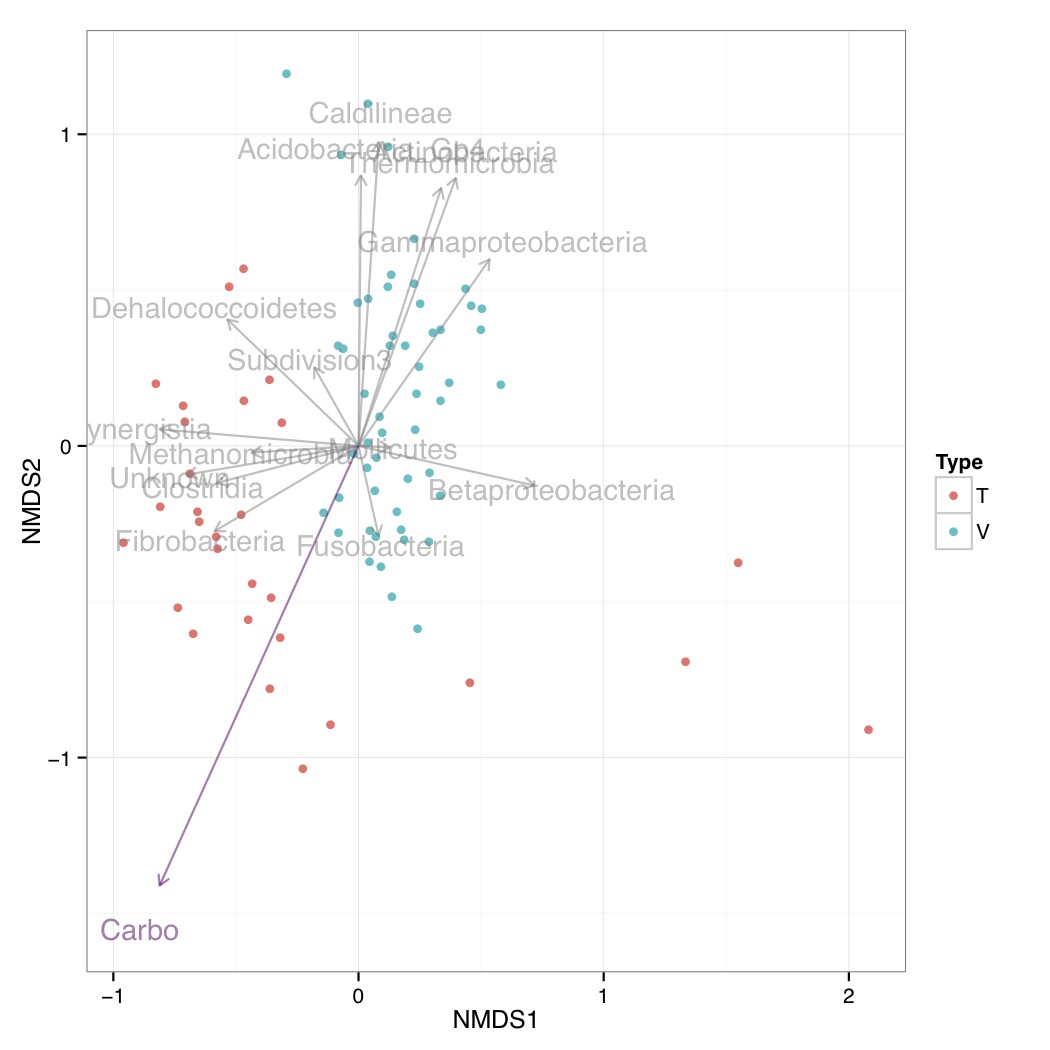
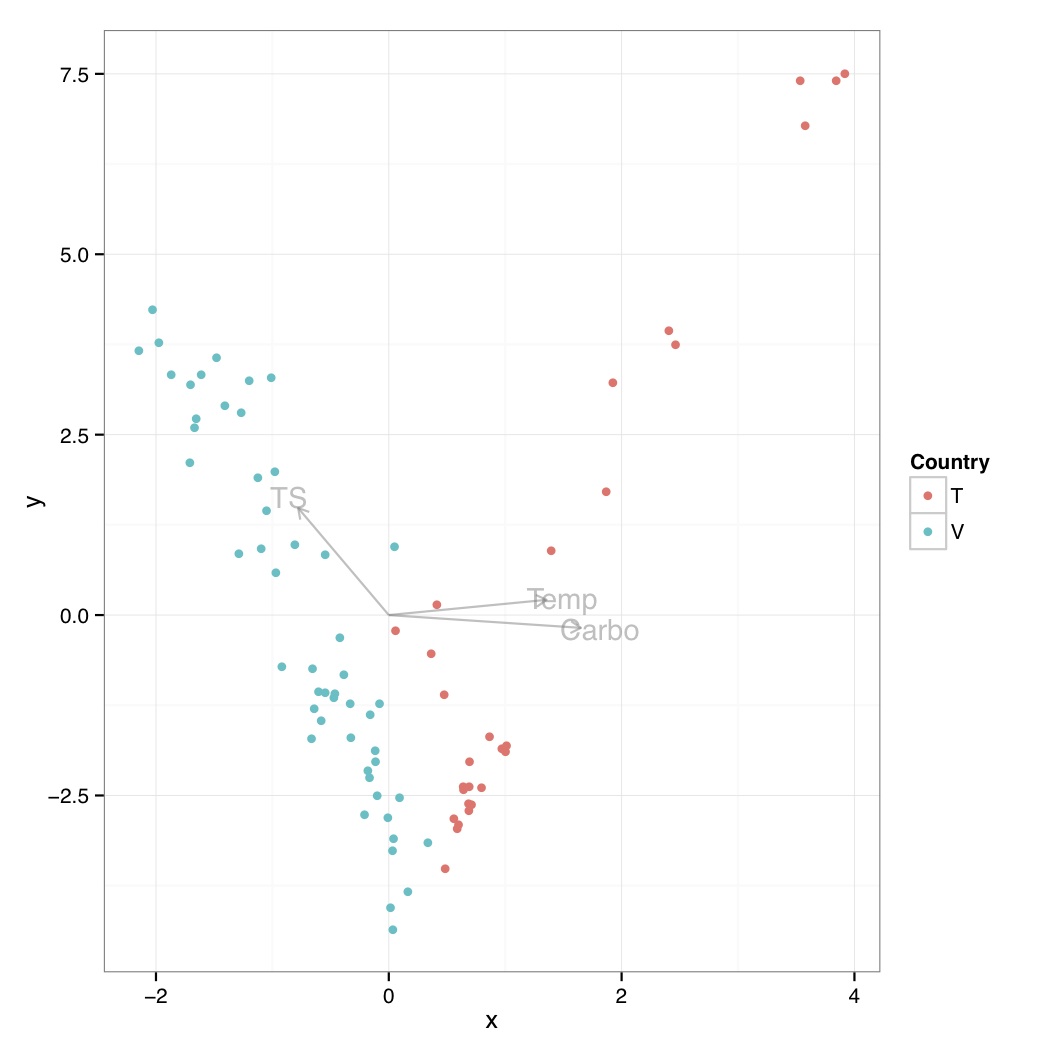
# ============================================================ # Tutorial on plotting significant taxa and environmental variables on an NMDS plot using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= library(vegan) library(ggplot2) library(grid) # This R script is an extension of vegan library's bioenv() # function and uses the bio.env() and bio.step() of # http://menugget.blogspot.co.uk/2011/06/clarke-and-ainsworths-bioenv-and-bvstep.html # The original author suggested these functions to overcome # the inflexibility of the bioenv() function which uses # a similarity matrix based on normalized "euclidean" distance. # The new functions are given below and implement the following algorithms: # Clarke, K. R & Ainsworth, M. 1993. A method of linking multivariate community structure to environmental variables. Marine Ecology Progress Series, 92, 205-219. # Clarke, K. R., Gorley, R. N., 2001. PRIMER v5: User Manual/Tutorial. PRIMER-E, Plymouth, UK. # Clarke, K. R., Warwick, R. M., 2001. Changes in Marine Communities: An Approach to Statistical Analysis and Interpretation, 2nd edition. PRIMER-E Ltd, Plymouth, UK. # Clarke, K. R., Warwick, R. M., 1998. Quantifying structural redundancy in ecological communities. Oecologia, 113:278-289. bv.step <- function(fix.mat, var.mat, fix.dist.method="bray", var.dist.method="euclidean", correlation.method="spearman", scale.fix=FALSE, scale.var=TRUE, max.rho=0.95, min.delta.rho=0.001, random.selection=TRUE, prop.selected.var=0.2, num.restarts=10, var.always.include=NULL, var.exclude=NULL, output.best=10 ){ if(dim(fix.mat)[1] != dim(var.mat)[1]){stop("fixed and variable matrices must have the same number of rows")} if(sum(var.always.include %in% var.exclude) > 0){stop("var.always.include and var.exclude share a variable")} require(vegan) if(scale.fix){fix.mat<-scale(fix.mat)}else{fix.mat<-fix.mat} if(scale.var){var.mat<-scale(var.mat)}else{var.mat<-var.mat} fix.dist <- vegdist(as.matrix(fix.mat), method=fix.dist.method) #an initial removal phase var.dist.full <- vegdist(as.matrix(var.mat), method=var.dist.method) full.cor <- suppressWarnings(cor.test(fix.dist, var.dist.full, method=correlation.method))$estimate var.comb <- combn(1:ncol(var.mat), ncol(var.mat)-1) RES <- data.frame(var.excl=rep(NA,ncol(var.comb)), n.var=ncol(var.mat)-1, rho=NA) for(i in 1:dim(var.comb)[2]){ var.dist <- vegdist(as.matrix(var.mat[,var.comb[,i]]), method=var.dist.method) temp <- suppressWarnings(cor.test(fix.dist, var.dist, method=correlation.method)) RES$var.excl[i] <- c(1:ncol(var.mat))[-var.comb[,i]] RES$rho[i] <- temp$estimate } delta.rho <- RES$rho - full.cor exclude <- sort(unique(c(RES$var.excl[which(abs(delta.rho) < min.delta.rho)], var.exclude))) if(random.selection){ num.restarts=num.restarts prop.selected.var=prop.selected.var prob<-rep(1,ncol(var.mat)) if(prop.selected.var< 1){ prob[exclude]<-0 } n.selected.var <- min(sum(prob),prop.selected.var*dim(var.mat)[2]) } else { num.restarts=1 prop.selected.var=1 prob<-rep(1,ncol(var.mat)) n.selected.var <- min(sum(prob),prop.selected.var*dim(var.mat)[2]) } RES_TOT <- c() for(i in 1:num.restarts){ step=1 RES <- data.frame(step=step, step.dir="F", var.incl=NA, n.var=0, rho=0) attr(RES$step.dir, "levels") <- c("F","B") best.comb <- which.max(RES$rho) best.rho <- RES$rho[best.comb] delta.rho <- Inf selected.var <- sort(unique(c(sample(1:dim(var.mat)[2], n.selected.var, prob=prob), var.always.include))) while(best.rho < max.rho & delta.rho > min.delta.rho & RES$n.var[best.comb] < length(selected.var)){ #forward step step.dir="F" step=step+1 var.comb <- combn(selected.var, RES$n.var[best.comb]+1, simplify=FALSE) if(RES$n.var[best.comb] == 0){ var.comb.incl<-1:length(var.comb) } else { var.keep <- as.numeric(unlist(strsplit(RES$var.incl[best.comb], ","))) temp <- NA*1:length(var.comb) for(j in 1:length(temp)){ temp[j] <- all(var.keep %in% var.comb[[j]]) } var.comb.incl <- which(temp==1) } RES.f <- data.frame(step=rep(step, length(var.comb.incl)), step.dir=step.dir, var.incl=NA, n.var=RES$n.var[best.comb]+1, rho=NA) for(f in 1:length(var.comb.incl)){ var.incl <- var.comb[[var.comb.incl[f]]] var.incl <- var.incl[order(var.incl)] var.dist <- vegdist(as.matrix(var.mat[,var.incl]), method=var.dist.method) temp <- suppressWarnings(cor.test(fix.dist, var.dist, method=correlation.method)) RES.f$var.incl[f] <- paste(var.incl, collapse=",") RES.f$rho[f] <- temp$estimate } last.F <- max(which(RES$step.dir=="F")) RES <- rbind(RES, RES.f[which.max(RES.f$rho),]) best.comb <- which.max(RES$rho) delta.rho <- RES$rho[best.comb] - best.rho best.rho <- RES$rho[best.comb] if(best.comb == step){ while(best.comb == step & RES$n.var[best.comb] > 1){ #backward step step.dir="B" step <- step+1 var.keep <- as.numeric(unlist(strsplit(RES$var.incl[best.comb], ","))) var.comb <- combn(var.keep, RES$n.var[best.comb]-1, simplify=FALSE) RES.b <- data.frame(step=rep(step, length(var.comb)), step.dir=step.dir, var.incl=NA, n.var=RES$n.var[best.comb]-1, rho=NA) for(b in 1:length(var.comb)){ var.incl <- var.comb[[b]] var.incl <- var.incl[order(var.incl)] var.dist <- vegdist(as.matrix(var.mat[,var.incl]), method=var.dist.method) temp <- suppressWarnings(cor.test(fix.dist, var.dist, method=correlation.method)) RES.b$var.incl[b] <- paste(var.incl, collapse=",") RES.b$rho[b] <- temp$estimate } RES <- rbind(RES, RES.b[which.max(RES.b$rho),]) best.comb <- which.max(RES$rho) best.rho<- RES$rho[best.comb] } } else { break() } } RES_TOT <- rbind(RES_TOT, RES[2:dim(RES)[1],]) print(paste(round((i/num.restarts)*100,3), "% finished")) } RES_TOT <- unique(RES_TOT[,3:5]) if(dim(RES_TOT)[1] > output.best){ order.by.best <- RES_TOT[order(RES_TOT$rho, decreasing=TRUE)[1:output.best],] } else { order.by.best <- RES_TOT[order(RES_TOT$rho, decreasing=TRUE), ] } rownames(order.by.best)<-NULL order.by.i.comb <- c() for(i in 1:length(selected.var)){ f1 <- which(RES_TOT$n.var==i) f2 <- which.max(RES_TOT$rho[f1]) order.by.i.comb <- rbind(order.by.i.comb, RES_TOT[f1[f2],]) } rownames(order.by.i.comb)<-NULL if(length(exclude)<1){var.exclude=NULL} else {var.exclude=exclude} out <- list( order.by.best=order.by.best, order.by.i.comb=order.by.i.comb, best.model.vars=paste(colnames(var.mat)[as.numeric(unlist(strsplit(order.by.best$var.incl[1], ",")))], collapse=","), best.model.rho=order.by.best$rho[1], var.always.include=var.always.include, var.exclude=var.exclude ) out } bio.env <- function(fix.mat, var.mat, fix.dist.method="bray", var.dist.method="euclidean", correlation.method="spearman", scale.fix=FALSE, scale.var=TRUE, output.best=10, var.max=ncol(var.mat) ){ if(dim(fix.mat)[1] != dim(var.mat)[1]){stop("fixed and variable matrices must have the same number of rows")} if(var.max > dim(var.mat)[2]){stop("var.max cannot be larger than the number of variables (columns) in var.mat")} require(vegan) combn.sum <- sum(factorial(ncol(var.mat))/(factorial(1:var.max)*factorial(ncol(var.mat)-1:var.max))) if(scale.fix){fix.mat<-scale(fix.mat)}else{fix.mat<-fix.mat} if(scale.var){var.mat<-scale(var.mat)}else{var.mat<-var.mat} fix.dist <- vegdist(fix.mat, method=fix.dist.method) RES_TOT <- c() best.i.comb <- c() iter <- 0 for(i in 1:var.max){ var.comb <- combn(1:ncol(var.mat), i, simplify=FALSE) RES <- data.frame(var.incl=rep(NA, length(var.comb)), n.var=i, rho=0) for(f in 1:length(var.comb)){ iter <- iter+1 var.dist <- vegdist(as.matrix(var.mat[,var.comb[[f]]]), method=var.dist.method) temp <- suppressWarnings(cor.test(fix.dist, var.dist, method=correlation.method)) RES$var.incl[f] <- paste(var.comb[[f]], collapse=",") RES$rho[f] <- temp$estimate if(iter %% 100 == 0){print(paste(round(iter/combn.sum*100, 3), "% finished"))} } order.rho <- order(RES$rho, decreasing=TRUE) best.i.comb <- c(best.i.comb, RES$var.incl[order.rho[1]]) if(length(order.rho) > output.best){ RES_TOT <- rbind(RES_TOT, RES[order.rho[1:output.best],]) } else { RES_TOT <- rbind(RES_TOT, RES) } } rownames(RES_TOT)<-NULL if(dim(RES_TOT)[1] > output.best){ order.by.best <- order(RES_TOT$rho, decreasing=TRUE)[1:output.best] } else { order.by.best <- order(RES_TOT$rho, decreasing=TRUE) } OBB <- RES_TOT[order.by.best,] rownames(OBB) <- NULL order.by.i.comb <- match(best.i.comb, RES_TOT$var.incl) OBC <- RES_TOT[order.by.i.comb,] rownames(OBC) <- NULL out <- list( order.by.best=OBB, order.by.i.comb=OBC, best.model.vars=paste(colnames(var.mat)[as.numeric(unlist(strsplit(OBB$var.incl[1], ",")))], collapse=",") , best.model.rho=OBB$rho[1] ) out } abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) meta_table<-read.csv("ENV_pitlatrine.csv",row.names=1,check.names=FALSE) #Just a check to ensure that the samples in meta_table are in the same order as in abund_table meta_table<-meta_table[rownames(abund_table),] #Get grouping information grouping_info<-data.frame(row.names=rownames(abund_table),t(as.data.frame(strsplit(rownames(abund_table),"_")))) # > head(grouping_info) # X1 X2 X3 # T_2_1 T 2 1 # T_2_10 T 2 10 # T_2_12 T 2 12 # T_2_2 T 2 2 # T_2_3 T 2 3 # T_2_6 T 2 6 #Parameters cmethod<-"pearson" #Correlation method to use: pearson, pearman, kendall fmethod<-"bray" #Fixed distance method: euclidean, manhattan, gower, altGower, canberra, bray, kulczynski, morisita,horn, binomial, and cao vmethod<-"bray" #Variable distance method: euclidean, manhattan, gower, altGower, canberra, bray, kulczynski, morisita,horn, binomial, and cao nmethod<-"bray" #NMDS distance method: euclidean, manhattan, gower, altGower, canberra, bray, kulczynski, morisita,horn, binomial, and cao res <- bio.env(wisconsin(abund_table), meta_table,fix.dist.method=fmethod, var.dist.method=vmethod, correlation.method=cmethod, scale.fix=FALSE, scale.var=TRUE) #Get the 10 best subset of environmental variables envNames<-colnames(meta_table) bestEnvFit<-"" for(i in (1:length(res$order.by.best$var.incl))) { bestEnvFit[i]<-paste(paste(envNames[as.numeric(unlist(strsplit(res$order.by.best$var.incl[i], split=",")))],collapse=' + '), " = ",res$order.by.best$rho[i],sep="") } bestEnvFit<-data.frame(bestEnvFit) colnames(bestEnvFit)<-"Best combination of environmental variables with similarity score" #> bestEnvFit #Best combination of environmental variables with similarity score #1 Carbo = 0.112933470129642 #2 Temp + VS + Carbo = 0.0850146558049758 #3 Temp + Carbo = 0.0836568099887996 #4 Temp = 0.0772024618792259 #5 perCODsbyt = 0.076360226020659 #6 TS + CODs + perCODsbyt + Prot + Carbo = 0.0752573390422113 #7 Temp + CODt + CODs + perCODsbyt + NH4 = 0.0749607646516179 #8 Temp + TS + VFA + perCODsbyt + NH4 + Prot + Carbo = 0.0718412745558854 #9 pH + Temp + TS + Carbo = 0.0693960850973541 #10 Temp + NH4 + Carbo = 0.0687961955200212 res.bv.step.biobio <- bv.step(wisconsin(abund_table), wisconsin(abund_table), fix.dist.method=fmethod, var.dist.method=vmethod,correlation.method=cmethod, scale.fix=FALSE, scale.var=FALSE, max.rho=0.95, min.delta.rho=0.001, random.selection=TRUE, prop.selected.var=0.3, num.restarts=10, output.best=10, var.always.include=NULL) #Get the 10 best subset of taxa taxaNames<-colnames(abund_table) bestTaxaFit<-"" for(i in (1:length(res.bv.step.biobio$order.by.best$var.incl))) { bestTaxaFit[i]<-paste(paste(taxaNames[as.numeric(unlist(strsplit(res.bv.step.biobio$order.by.best$var.incl[i], split=",")))],collapse=' + '), " = ",res.bv.step.biobio$order.by.best$rho[i],sep="") } bestTaxaFit<-data.frame(bestTaxaFit) colnames(bestTaxaFit)<-"Best combination of taxa with similarity score" #> bestTaxaFit #Best combination of taxa with similarity score #1 Bacilli + Bacteroidia + Chrysiogenetes + Clostridia + Dehalococcoidetes + Fibrobacteria + Flavobacteria + Fusobacteria + Gammaproteobacteria + Methanomicrobia + Mollicutes + Opitutae + Synergistia + Thermomicrobia + Unknown = 0.900998476657462 #2 Bacilli + Bacteroidia + Clostridia + Dehalococcoidetes + Fibrobacteria + Flavobacteria + Fusobacteria + Gammaproteobacteria + Methanomicrobia + Mollicutes + Opitutae + Synergistia + Thermomicrobia + Unknown = 0.899912718316228 #3 Bacilli + Bacteroidia + Clostridia + Dehalococcoidetes + Fibrobacteria + Flavobacteria + Fusobacteria + Gammaproteobacteria + Methanomicrobia + Mollicutes + Opitutae + Synergistia + Thermomicrobia = 0.896821772937576 #4 Clostridia + Dehalococcoidetes + Deltaproteobacteria + Epsilonproteobacteria + Erysipelotrichi + Flavobacteria + Fusobacteria + Methanobacteria + Mollicutes + Opitutae + Planctomycetacia + Sphingobacteria + Subdivision3 + Synergistia + Thermomicrobia = 0.892670058822226 #5 Bacilli + Bacteroidia + Clostridia + Dehalococcoidetes + Fibrobacteria + Flavobacteria + Fusobacteria + Gammaproteobacteria + Methanomicrobia + Opitutae + Synergistia + Thermomicrobia = 0.892533063335985 #6 Clostridia + Dehalococcoidetes + Deltaproteobacteria + Epsilonproteobacteria + Erysipelotrichi + Flavobacteria + Fusobacteria + Methanobacteria + Mollicutes + Opitutae + Planctomycetacia + Sphingobacteria + Subdivision3 + Synergistia = 0.891217789278463 #7 Clostridia + Dehalococcoidetes + Deltaproteobacteria + Epsilonproteobacteria + Erysipelotrichi + Flavobacteria + Fusobacteria + Mollicutes + Opitutae + Planctomycetacia + Sphingobacteria + Subdivision3 + Synergistia = 0.888669881927483 #8 Bacilli + Bacteroidia + Clostridia + Dehalococcoidetes + Fibrobacteria + Flavobacteria + Fusobacteria + Gammaproteobacteria + Opitutae + Synergistia + Thermomicrobia = 0.887052815492516 #9 Clostridia + Dehalococcoidetes + Deltaproteobacteria + Epsilonproteobacteria + Erysipelotrichi + Flavobacteria + Fusobacteria + Opitutae + Planctomycetacia + Sphingobacteria + Subdivision3 + Synergistia = 0.885880090785632 #10 Acidobacteria_Gp4 + Actinobacteria + Bacilli + Betaproteobacteria + Clostridia + Dehalococcoidetes + Erysipelotrichi + Fibrobacteria + Lentisphaeria + Methanomicrobia + Opitutae + Sphingobacteria + Thermomicrobia + Unknown = 0.882956559638505 #Generate NMDS plot MDS_res=metaMDS(abund_table, distance = nmethod, k = 2, trymax = 50) bio.keep <- as.numeric(unlist(strsplit(res.bv.step.biobio$order.by.best$var.incl[1], ","))) bio.fit <- envfit(MDS_res, abund_table[,bio.keep,drop=F], perm = 999) #> bio.fit # #***VECTORS # #NMDS1 NMDS2 r2 Pr(>r) #Bacilli 0.94632 0.32322 0.0423 0.167 #Bacteroidia -0.24478 -0.96958 0.0383 0.218 #Chrysiogenetes -0.22674 0.97395 0.0078 0.660 #Clostridia -0.98067 -0.19567 0.1092 0.017 * #Dehalococcoidetes -0.79160 0.61103 0.1421 0.009 ** #Fibrobacteria -0.90885 -0.41712 0.1320 0.010 ** #Flavobacteria 0.56629 0.82421 0.1746 0.003 ** #Fusobacteria 0.26601 -0.96397 0.0284 0.333 #Gammaproteobacteria 0.67008 0.74229 0.2024 0.001 *** #Methanomicrobia -0.99912 -0.04188 0.0602 0.120 #Mollicutes 0.99885 -0.04798 0.0052 0.769 #Opitutae 0.55774 0.83002 0.2033 0.002 ** #Synergistia -0.99732 0.07322 0.2079 0.002 ** #Thermomicrobia 0.38169 0.92429 0.2514 0.001 *** #Unknown -0.99232 -0.12373 0.1570 0.009 ** #--- #Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 #Permutation: free #Number of permutations: 999 #use the best set of environmental variables in env.keep eval(parse(text=paste("env.keep <- c(",res$order.by.best$var.incl[1],")",sep=""))) env.fit <- envfit(MDS_res, meta_table[,env.keep,drop=F], perm = 999) #Get site information df<-scores(MDS_res,display=c("sites")) #Add grouping information df<-data.frame(df,Type=grouping_info[rownames(df),1]) #Get the vectors for bioenv.fit df_biofit<-scores(bio.fit,display=c("vectors")) df_biofit<-df_biofit*vegan:::ordiArrowMul(df_biofit) df_biofit<-as.data.frame(df_biofit) #Get the vectors for env.fit df_envfit<-scores(env.fit,display=c("vectors")) df_envfit<-df_envfit*vegan:::ordiArrowMul(df_envfit) df_envfit<-as.data.frame(df_envfit) #Draw samples p<-ggplot() p<-p+geom_point(data=df,aes(NMDS1,NMDS2,colour=Type)) #Draw taxas p<-p+geom_segment(data=df_biofit, aes(x = 0, y = 0, xend = NMDS1, yend = NMDS2), arrow = arrow(length = unit(0.2, "cm")),color="#808080",alpha=0.5) p<-p+geom_text(data=as.data.frame(df_biofit*1.1),aes(NMDS1, NMDS2, label = rownames(df_biofit)),color="#808080",alpha=0.5) #Draw environmental variables p<-p+geom_segment(data=df_envfit, aes(x = 0, y = 0, xend = NMDS1, yend = NMDS2), arrow = arrow(length = unit(0.2, "cm")),color="#4C005C",alpha=0.5) p<-p+geom_text(data=as.data.frame(df_envfit*1.1),aes(NMDS1, NMDS2, label = rownames(df_envfit)),color="#4C005C",alpha=0.5) p<-p+theme_bw() pdf("NMDS_bioenv.pdf") print(p) dev.off()
# ============================================================ # Tutorial on drawing a CCA plot with significant environmental variables using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= library(vegan) library(grid) abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) meta_table<-read.csv("ENV_pitlatrine.csv",row.names=1,check.names=FALSE) #Just a check to ensure that the samples in meta_table are in the same order as in abund_table meta_table<-meta_table[rownames(abund_table),] #Filter out any samples taxas that have zero entries abund_table<-subset(abund_table,rowSums(abund_table)!=0) #Convert to relative frequencies abund_table<-abund_table/rowSums(abund_table) #Use adonis to find significant environmental variables abund_table.adonis <- adonis(abund_table ~ ., data=meta_table) #> abund_table.adonis #Call: # adonis(formula = abund_table ~ ., data = meta_table) #Permutation: free #Number of permutations: 999 # #Terms added sequentially (first to last) # #Df SumsOfSqs MeanSqs F.Model R2 Pr(>F) #pH 1 0.2600 0.26002 2.1347 0.01974 0.067 . #Temp 1 1.6542 1.65417 13.5806 0.12559 0.001 *** #TS 1 0.8298 0.82983 6.8128 0.06300 0.001 *** #VS 1 0.3143 0.31434 2.5807 0.02387 0.038 * #VFA 1 0.2280 0.22796 1.8715 0.01731 0.109 #CODt 1 0.1740 0.17400 1.4285 0.01321 0.209 #CODs 1 0.0350 0.03504 0.2877 0.00266 0.909 #perCODsbyt 1 0.1999 0.19986 1.6409 0.01517 0.148 #NH4 1 0.1615 0.16154 1.3262 0.01226 0.236 #Prot 1 0.3657 0.36570 3.0024 0.02777 0.024 * #Carbo 1 0.5444 0.54439 4.4693 0.04133 0.003 ** #Residuals 69 8.4045 0.12180 0.63809 #Total 80 13.1713 1.00000 #--- # Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 #Extract the best variables bestEnvVariables<-rownames(abund_table.adonis$aov.tab)[abund_table.adonis$aov.tab$"Pr(>F)"<=0.01] #Last two are NA entries, so we have to remove them bestEnvVariables<-bestEnvVariables[!is.na(bestEnvVariables)] #We are now going to use only those environmental variables in cca that were found significant eval(parse(text=paste("sol <- cca(abund_table ~ ",do.call(paste,c(as.list(bestEnvVariables),sep=" + ")),",data=meta_table)",sep=""))) #You can use the following to use all the environmental variables #sol<-cca(abund_table ~ ., data=meta_table) scrs<-scores(sol,display=c("sp","wa","lc","bp","cn")) #Check the attributes # > attributes(scrs) # $names # [1] "species" "sites" "constraints" "biplot" # [5] "centroids" #Extract site data first df_sites<-data.frame(scrs$sites,t(as.data.frame(strsplit(rownames(scrs$sites),"_")))) colnames(df_sites)<-c("x","y","Country","Latrine","Depth") #Draw sites p<-ggplot() p<-p+geom_point(data=df_sites,aes(x,y,colour=Country)) #Draw biplots multiplier <- vegan:::ordiArrowMul(scrs$biplot) # Reference: http://www.inside-r.org/packages/cran/vegan/docs/envfit # The printed output of continuous variables (vectors) gives the direction cosines # which are the coordinates of the heads of unit length vectors. In plot these are # scaled by their correlation (square root of the column r2) so that "weak" predictors # have shorter arrows than "strong" predictors. You can see the scaled relative lengths # using command scores. The plotted (and scaled) arrows are further adjusted to the # current graph using a constant multiplier: this will keep the relative r2-scaled # lengths of the arrows but tries to fill the current plot. You can see the multiplier # using vegan:::ordiArrowMul(result_of_envfit), and set it with the argument arrow.mul. df_arrows<- scrs$biplot*multiplier colnames(df_arrows)<-c("x","y") df_arrows=as.data.frame(df_arrows) p<-p+geom_segment(data=df_arrows, aes(x = 0, y = 0, xend = x, yend = y), arrow = arrow(length = unit(0.2, "cm")),color="#808080",alpha=0.5) p<-p+geom_text(data=as.data.frame(df_arrows*1.1),aes(x, y, label = rownames(df_arrows)),color="#808080",alpha=0.5) # Draw species df_species<- as.data.frame(scrs$species) colnames(df_species)<-c("x","y") # Either choose text or points #p<-p+geom_text(data=df_species,aes(x,y,label=rownames(df_species))) #p<-p+geom_point(data=df_species,aes(x,y,shape="Species"))+scale_shape_manual("",values=2) p<-p+theme_bw() pdf("CCA.pdf") print(p) dev.off()
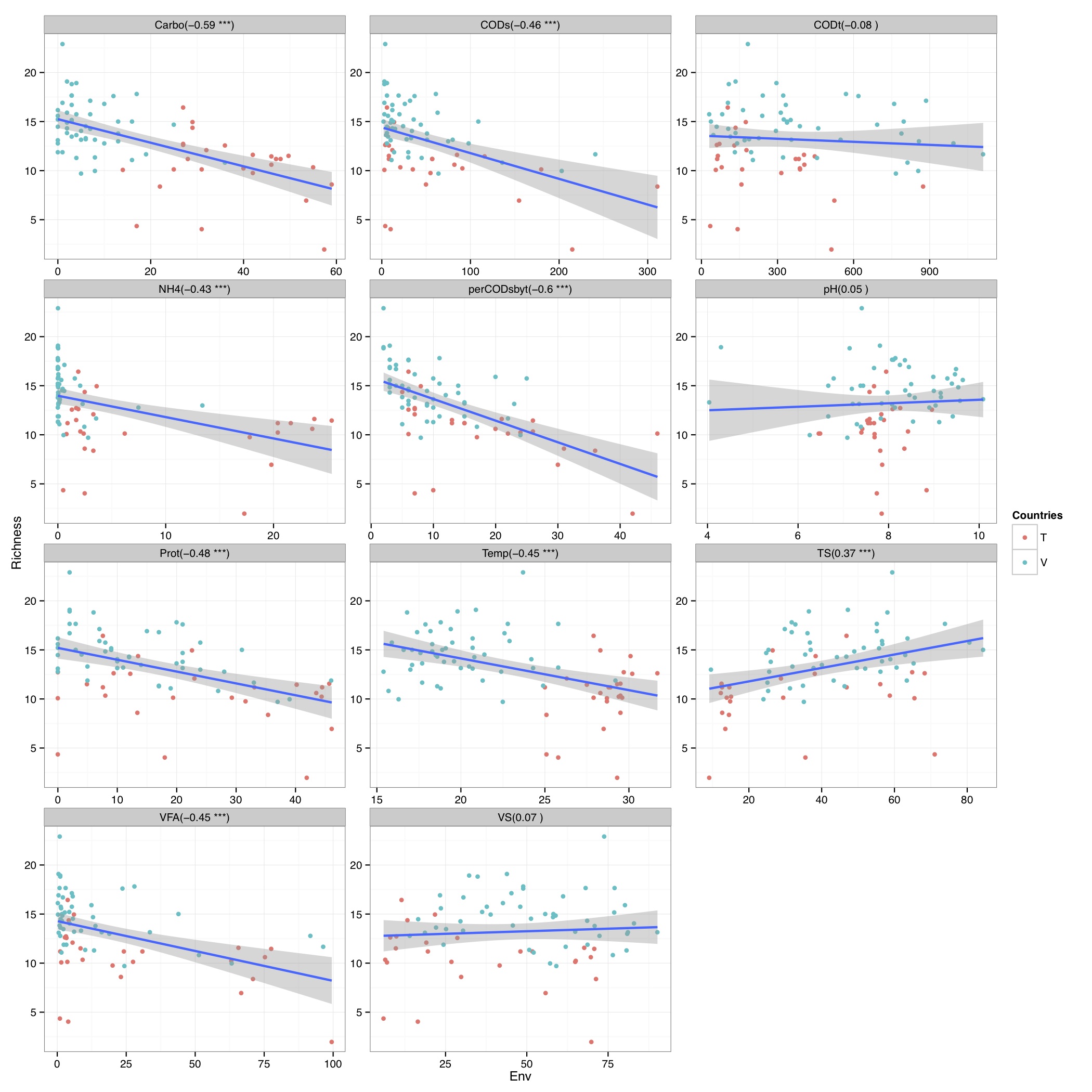
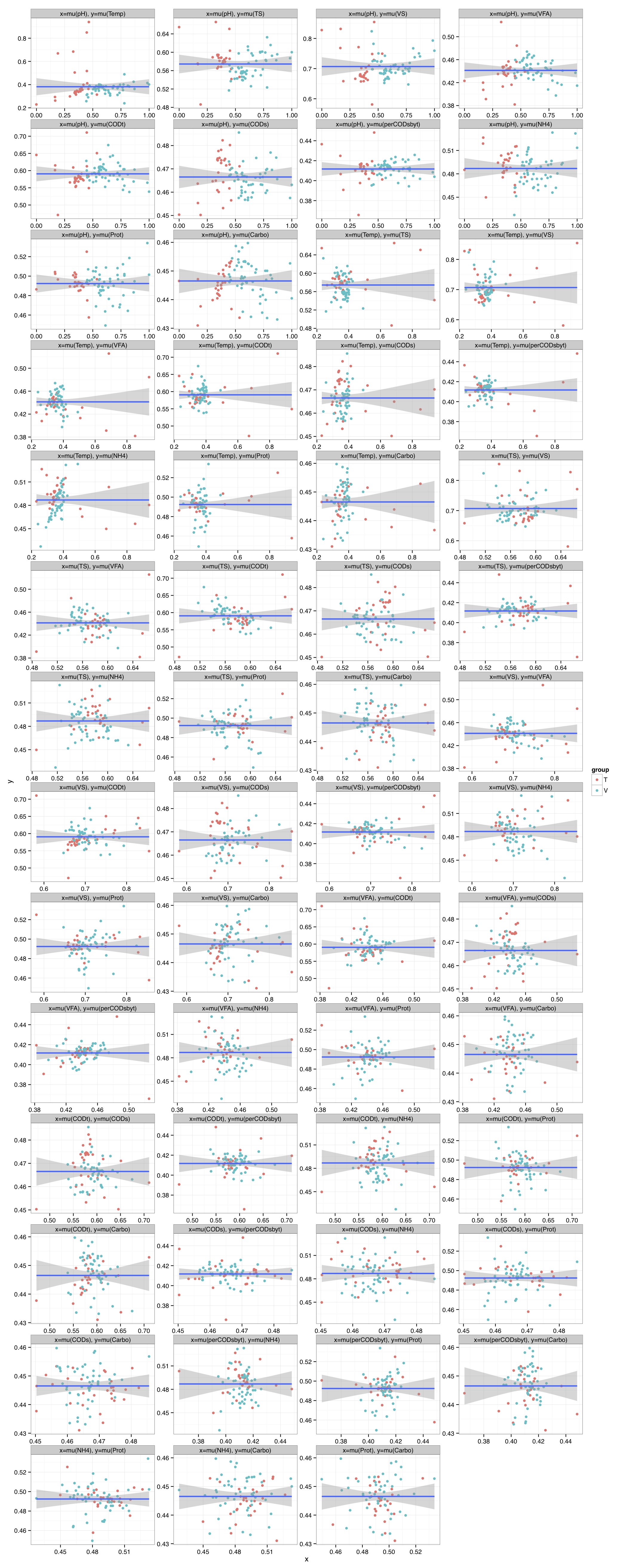
# ============================================================ # Tutorial on drawing a richness plot using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) meta_table<-read.csv("ENV_pitlatrine.csv",row.names=1,check.names=FALSE) #Filter out samples with fewer counts abund_table<-abund_table[rowSums(abund_table)>200,] #Extract the corresponding meta_table for the samples in abund_table meta_table<-meta_table[rownames(abund_table),] #Load vegan library library(vegan) # Calculate species richness N <- rowSums(abund_table) S <- specnumber(abund_table) S.rar <-rarefy(abund_table, min(N)) # Regression of S.rar against meta_table S.lm <- lm(S.rar ~ ., data = meta_table) summary(S.lm) #Format the data for ggplot df_S<-NULL for (i in 1:dim(meta_table)[2]){ tmp<-data.frame(row.names=NULL,Sample=names(S.rar),Richness=S.rar,Env=meta_table[,i],Label=rep(colnames(meta_table)[i],length(meta_table[,i]))) if (is.null(df_S)){ df_S=tmp }else{ df_S<-rbind(df_S,tmp) } } #Get grouping information grouping_info<-data.frame(row.names=rownames(df_S),t(as.data.frame(strsplit(as.character(df_S[,"Sample"]),"_")))) colnames(grouping_info)<-c("Countries","Latrine","Depth") #Merge this information with df_S df_S<-cbind(df_S,grouping_info) # > head(df_S) # Sample Richness Env Label Countries Latrine Depth # 1 T_2_1 8.424693 7.82 pH T 2 1 # 2 T_2_12 4.389534 8.84 pH T 2 12 # 3 T_2_2 10.141087 6.49 pH T 2 2 # 4 T_2_3 10.156289 6.46 pH T 2 3 # 5 T_2_6 10.120478 7.69 pH T 2 6 # 6 T_2_9 11.217727 7.60 pH T 2 9 #We now make sure there are no factors in df_S df_S$Label<-as.character(df_S$Label) df_S$Countries<-as.character(df_S$Countries) df_S$Latrine<-as.character(df_S$Latrine) df_S$Depth<-as.character(df_S$Depth) #We need a Pvalue formatter formatPvalues <- function(pvalue) { ra<-"" if(pvalue <= 0.1) ra<-"." if(pvalue <= 0.05) ra<-"*" if(pvalue <= 0.01) ra<-"**" if(pvalue <= 0.001) ra<-"***" return(ra) } #Now use ddply to get correlations library(plyr) cors<-ddply(df_S,.(Label), summarise, cor=round(cor(Richness,Env),2), sig=formatPvalues(cor.test(Richness,Env)$p.value)) # > head(cors) # Label cor sig # 1 Carbo -0.59 *** # 2 CODs -0.46 *** # 3 CODt -0.08 # 4 NH4 -0.43 *** # 5 perCODsbyt -0.60 *** # 6 pH 0.05 #We have to do a trick to assign labels as facet_wrap doesn't have an explicit labeller #Reference 1: http://stackoverflow.com/questions/19282897/how-to-add-expressions-to-labels-in-facet-wrap #Reference 2: http://stackoverflow.com/questions/11979017/changing-facet-label-to-math-formula-in-ggplot2 facet_wrap_labeller <- function(gg.plot,labels=NULL) { #works with R 3.0.1 and ggplot2 0.9.3.1 require(gridExtra) g <- ggplotGrob(gg.plot) gg <- g$grobs strips <- grep("strip_t", names(gg)) for(ii in seq_along(labels)) { modgrob <- getGrob(gg[[strips[ii]]], "strip.text", grep=TRUE, global=TRUE) gg[[strips[ii]]]$children[[modgrob$name]] <- editGrob(modgrob,label=labels[ii]) } g$grobs <- gg class(g) = c("arrange", "ggplot",class(g)) g } #Now convert your labels wrap_labels<-do.call(paste,c(cors[c("Label")],"(",cors[c("cor")]," ",cors[c("sig")],")",sep="")) p<-ggplot(df_S,aes(Env,Richness)) + geom_point(aes(colour=Countries)) + geom_smooth(,method="lm", size=1, se=T) + facet_wrap( ~ Label , scales="free", ncol=3) +theme_bw() p<-facet_wrap_labeller(p,labels=wrap_labels) pdf("Richness.pdf",width=14,height=14) print(p) dev.off()
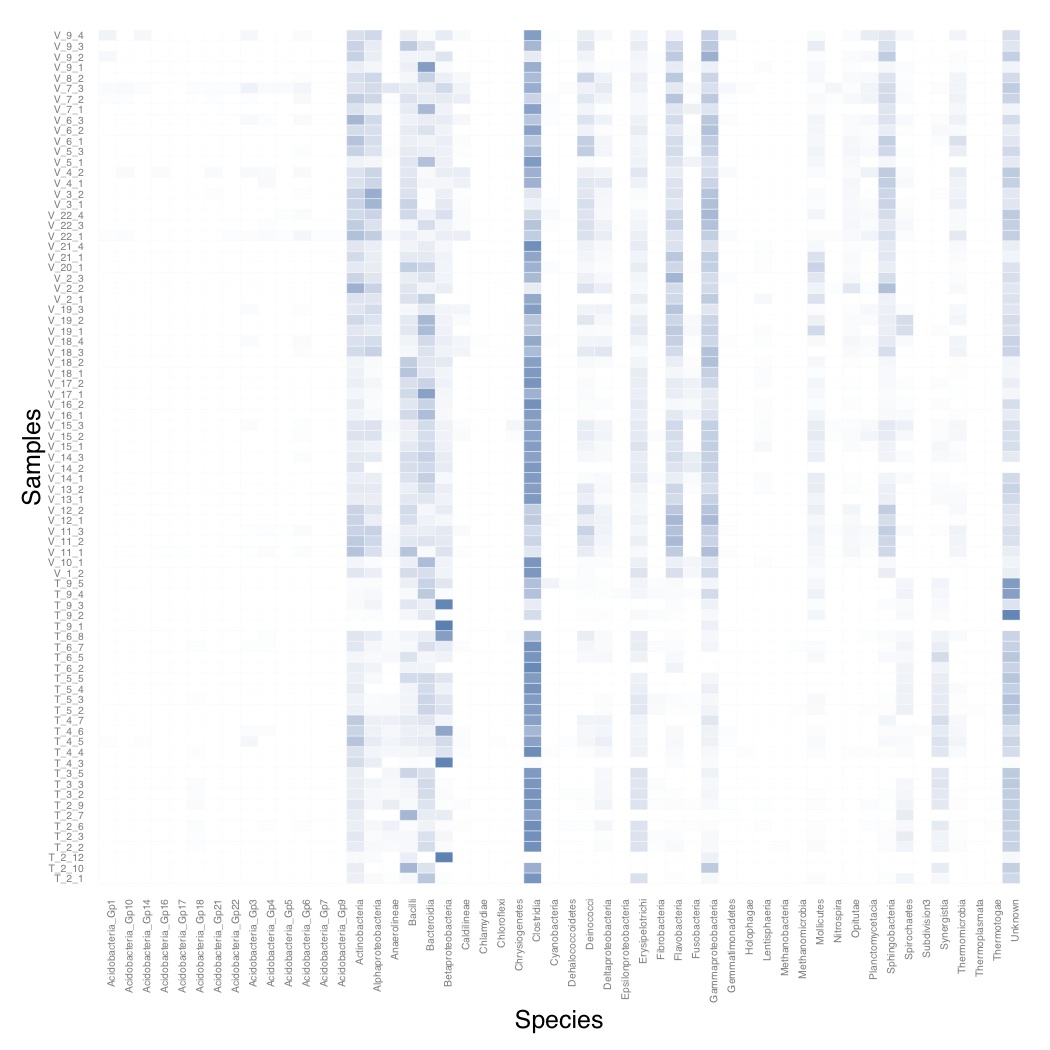
# ============================================================ # Tutorial on drawing a heatmap using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) # Transpose the data to have sample names on rows abund_table<-t(abund_table) # Convert to relative frequencies abund_table <- abund_table/rowSums(abund_table) library(reshape2) df<-melt(abund_table) colnames(df)<-c("Samples","Species","Value") library(plyr) library(scales) # We are going to apply transformation to our data to make it # easier on eyes #df<-ddply(df,.(Samples),transform,rescale=scale(Value)) df<-ddply(df,.(Samples),transform,rescale=sqrt(Value)) # Plot heatmap p <- ggplot(df, aes(Species, Samples)) + geom_tile(aes(fill = rescale),colour = "white") + scale_fill_gradient(low = "white",high = "steelblue")+ scale_x_discrete(expand = c(0, 0)) + scale_y_discrete(expand = c(0, 0)) + theme(legend.position = "none",axis.ticks = element_blank(),axis.text.x = element_text(angle = 90, hjust = 1,size=5),axis.text.y = element_text(size=5)) pdf("Heatmap.pdf") print(p) dev.off()
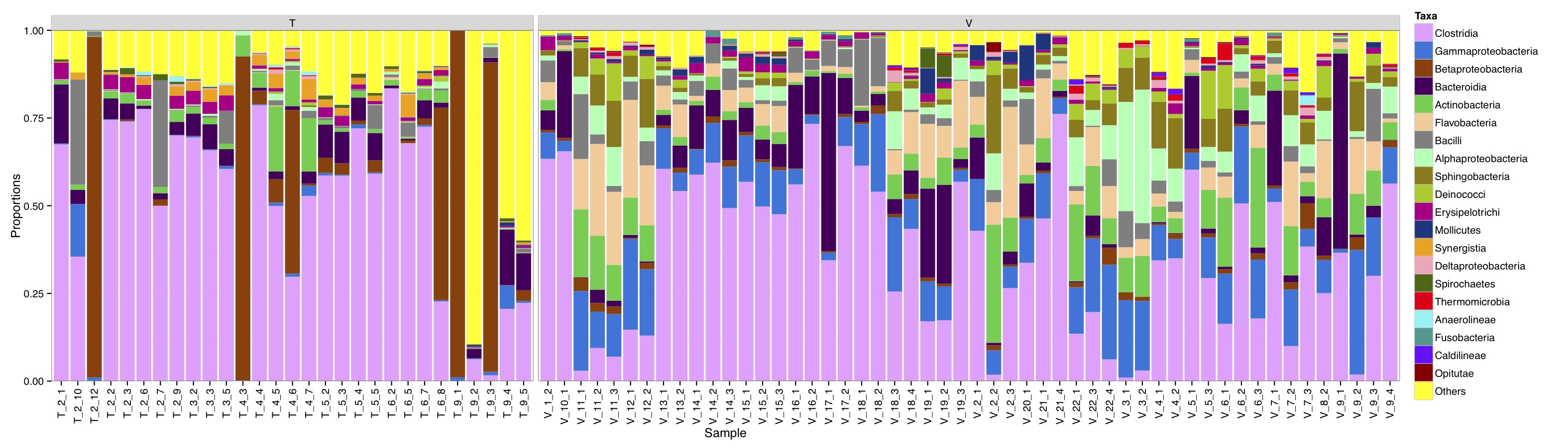
# ============================================================ # Tutorial on drawing a taxa plot using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) meta_table<-read.csv("ENV_pitlatrine.csv",row.names=1,check.names=FALSE) #Just a check to ensure that the samples in meta_table are in the same order as in abund_table meta_table<-meta_table[rownames(abund_table),] #Get grouping information grouping_info<-data.frame(row.names=rownames(abund_table),t(as.data.frame(strsplit(rownames(abund_table),"_")))) # > head(grouping_info) # X1 X2 X3 # T_2_1 T 2 1 # T_2_10 T 2 10 # T_2_12 T 2 12 # T_2_2 T 2 2 # T_2_3 T 2 3 # T_2_6 T 2 6 #Apply proportion normalisation x<-abund_table/rowSums(abund_table) x<-x[,order(colSums(x),decreasing=TRUE)] #Extract list of top N Taxa N<-21 taxa_list<-colnames(x)[1:N] #remove "__Unknown__" and add it to others taxa_list<-taxa_list[!grepl("Unknown",taxa_list)] N<-length(taxa_list) #Generate a new table with everything added to Others new_x<-data.frame(x[,colnames(x) %in% taxa_list],Others=rowSums(x[,!colnames(x) %in% taxa_list])) #You can change the Type=grouping_info[,1] should you desire any other grouping of panels df<-NULL for (i in 1:dim(new_x)[2]){ tmp<-data.frame(row.names=NULL,Sample=rownames(new_x),Taxa=rep(colnames(new_x)[i],dim(new_x)[1]),Value=new_x[,i],Type=grouping_info[,1]) if(i==1){df<-tmp} else {df<-rbind(df,tmp)} } colours <- c("#F0A3FF", "#0075DC", "#993F00","#4C005C","#2BCE48","#FFCC99","#808080","#94FFB5","#8F7C00","#9DCC00","#C20088","#003380","#FFA405","#FFA8BB","#426600","#FF0010","#5EF1F2","#00998F","#740AFF","#990000","#FFFF00"); library(ggplot2) p<-ggplot(df,aes(Sample,Value,fill=Taxa))+geom_bar(stat="identity")+facet_grid(. ~ Type, drop=TRUE,scale="free",space="free_x") p<-p+scale_fill_manual(values=colours[1:(N+1)]) p<-p+theme_bw()+ylab("Proportions") p<-p+ scale_y_continuous(expand = c(0,0))+theme(strip.background = element_rect(fill="gray85"))+theme(panel.margin = unit(0.3, "lines")) p<-p+theme(axis.text.x=element_text(angle=90,hjust=1,vjust=0.5)) pdf("TAXAplot.pdf",height=6,width=21) print(p) dev.off()
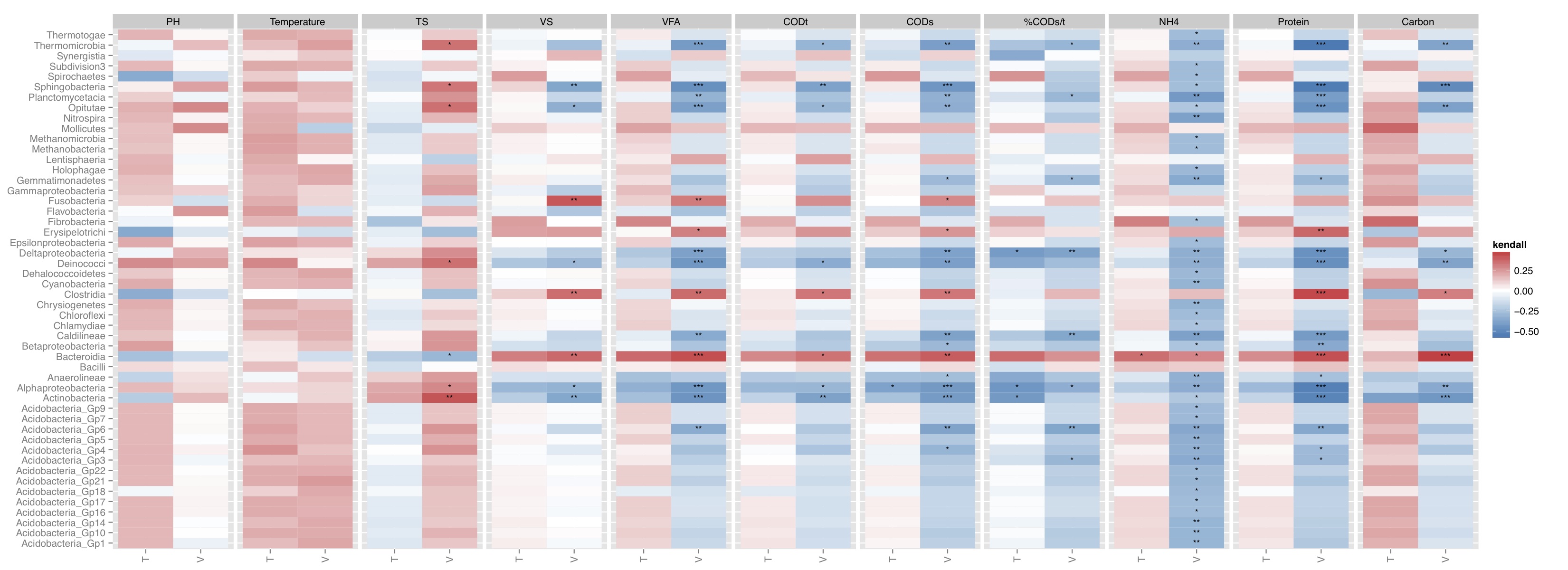
# ============================================================ # Tutorial on drawing a correlation map using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) meta_table<-read.csv("ENV_pitlatrine.csv",row.names=1,check.names=FALSE) #Filter out samples with fewer counts abund_table<-abund_table[rowSums(abund_table)>200,] #Extract the corresponding meta_table for the samples in abund_table meta_table<-meta_table[rownames(abund_table),] #You can use sel_env to specify the variables you want to use and sel_env_label to specify the labes for the pannel sel_env<-c("pH","Temp","TS","VS","VFA","CODt","CODs","perCODsbyt","NH4","Prot","Carbo") sel_env_label <- list( 'pH'="PH", 'Temp'="Temperature", 'TS'="TS", 'VS'="VS", 'VFA'="VFA", 'CODt'="CODt", 'CODs'="CODs", 'perCODsbyt'="%CODs/t", 'NH4'="NH4", 'Prot'="Protein", 'Carbo'="Carbon" ) sel_env_label<-t(as.data.frame(sel_env_label)) sel_env_label<-as.data.frame(sel_env_label) colnames(sel_env_label)<-c("Trans") sel_env_label$Trans<-as.character(sel_env_label$Trans) #Now get a filtered table based on sel_env meta_table_filtered<-meta_table[,sel_env] abund_table_filtered<-abund_table[rownames(meta_table_filtered),] #Apply normalisation (either use relative or log-relative transformation) #x<-abund_table_filtered/rowSums(abund_table_filtered) x<-log((abund_table_filtered+1)/(rowSums(abund_table_filtered)+dim(abund_table_filtered)[2])) x<-x[,order(colSums(x),decreasing=TRUE)] #Extract list of top N Taxa N<-51 taxa_list<-colnames(x)[1:N] #remove "__Unknown__" and add it to others taxa_list<-taxa_list[!grepl("Unknown",taxa_list)] N<-length(taxa_list) x<-data.frame(x[,colnames(x) %in% taxa_list]) y<-meta_table_filtered #Get grouping information grouping_info<-data.frame(row.names=rownames(abund_table),t(as.data.frame(strsplit(rownames(abund_table),"_")))) # > head(grouping_info) # X1 X2 X3 # T_2_1 T 2 1 # T_2_10 T 2 10 # T_2_12 T 2 12 # T_2_2 T 2 2 # T_2_3 T 2 3 # T_2_6 T 2 6 #Let us group on countries groups<-grouping_info[,1] #You can use kendall, spearman, or pearson below: method<-"kendall" #Now calculate the correlation between individual Taxa and the environmental data df<-NULL for(i in colnames(x)){ for(j in colnames(y)){ for(k in unique(groups)){ a<-x[groups==k,i,drop=F] b<-y[groups==k,j,drop=F] tmp<-c(i,j,cor(a[complete.cases(b),],b[complete.cases(b),],use="everything",method=method),cor.test(a[complete.cases(b),],b[complete.cases(b),],method=method)$p.value,k) if(is.null(df)){ df<-tmp } else{ df<-rbind(df,tmp) } } } } df<-data.frame(row.names=NULL,df) colnames(df)<-c("Taxa","Env","Correlation","Pvalue","Type") df$Pvalue<-as.numeric(as.character(df$Pvalue)) df$AdjPvalue<-rep(0,dim(df)[1]) df$Correlation<-as.numeric(as.character(df$Correlation)) #You can adjust the p-values for multiple comparison using Benjamini & Hochberg (1995): # 1 -> donot adjust # 2 -> adjust Env + Type (column on the correlation plot) # 3 -> adjust Taxa + Type (row on the correlation plot for each type) # 4 -> adjust Taxa (row on the correlation plot) # 5 -> adjust Env (panel on the correlation plot) adjustment_label<-c("NoAdj","AdjEnvAndType","AdjTaxaAndType","AdjTaxa","AdjEnv") adjustment<-5 if(adjustment==1){ df$AdjPvalue<-df$Pvalue } else if (adjustment==2){ for(i in unique(df$Env)){ for(j in unique(df$Type)){ sel<-df$Env==i & df$Type==j df$AdjPvalue[sel]<-p.adjust(df$Pvalue[sel],method="BH") } } } else if (adjustment==3){ for(i in unique(df$Taxa)){ for(j in unique(df$Type)){ sel<-df$Taxa==i & df$Type==j df$AdjPvalue[sel]<-p.adjust(df$Pvalue[sel],method="BH") } } } else if (adjustment==4){ for(i in unique(df$Taxa)){ sel<-df$Taxa==i df$AdjPvalue[sel]<-p.adjust(df$Pvalue[sel],method="BH") } } else if (adjustment==5){ for(i in unique(df$Env)){ sel<-df$Env==i df$AdjPvalue[sel]<-p.adjust(df$Pvalue[sel],method="BH") } } #Now we generate the labels for signifant values df$Significance<-cut(df$AdjPvalue, breaks=c(-Inf, 0.001, 0.01, 0.05, Inf), label=c("***", "**", "*", "")) #We ignore NAs df<-df[complete.cases(df),] #We want to reorganize the Env data based on they appear df$Env<-factor(df$Env,as.character(df$Env)) #We use the function to change the labels for facet_grid in ggplot2 Env_labeller <- function(variable,value){ return(sel_env_label[as.character(value),"Trans"]) } p <- ggplot(aes(x=Type, y=Taxa, fill=Correlation), data=df) p <- p + geom_tile() + scale_fill_gradient2(low="#2C7BB6", mid="white", high="#D7191C") p<-p+theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust=0.5)) p<-p+geom_text(aes(label=Significance), color="black", size=3)+labs(y=NULL, x=NULL, fill=method) p<-p+facet_grid(. ~ Env, drop=TRUE,scale="free",space="free_x",labeller=Env_labeller) pdf(paste("Correlation_",adjustment_label[adjustment],".pdf",sep=""),height=8,width=22) print(p) dev.off()
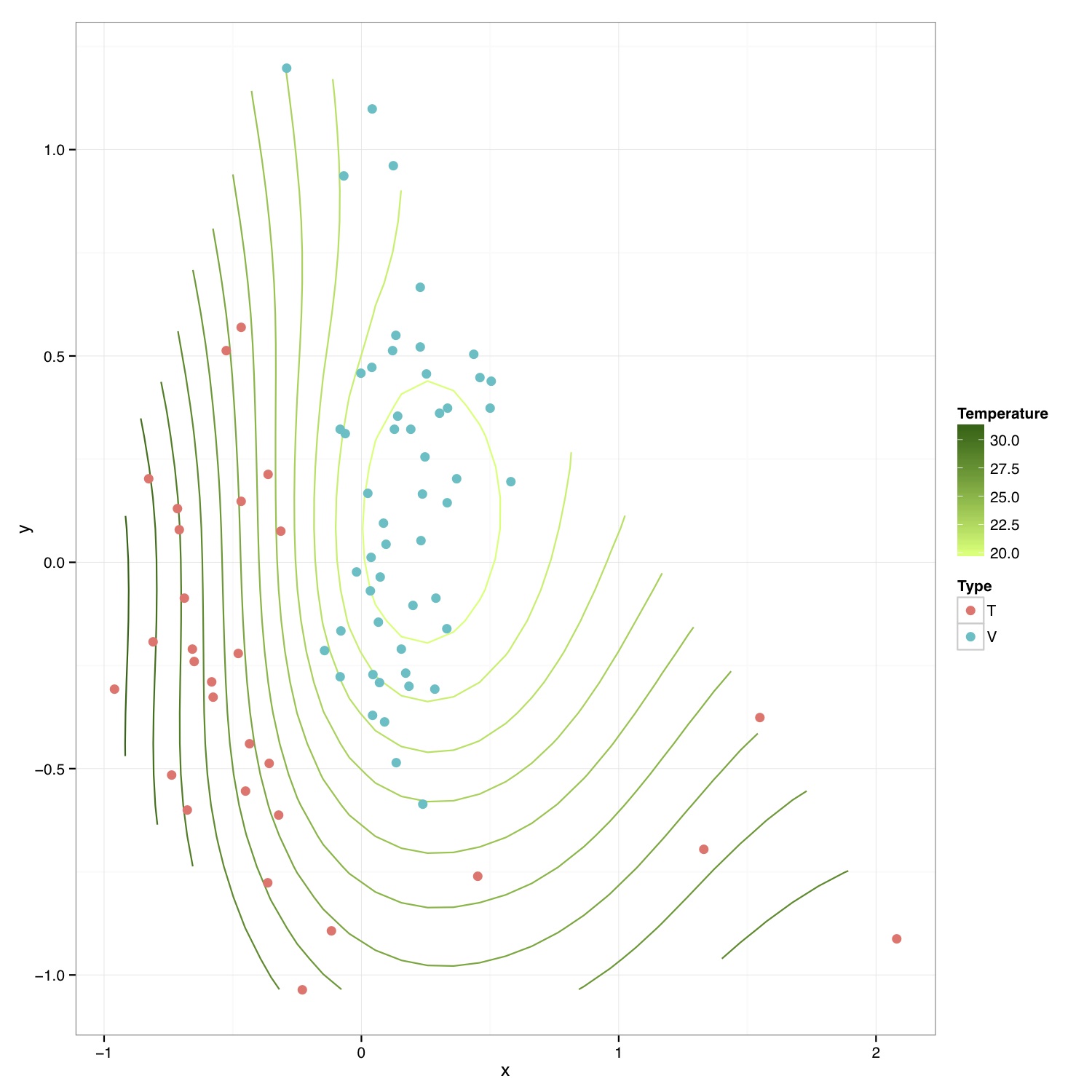
# ============================================================ # Tutorial on drawing an NMDS plot with environmental contours using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) meta_table<-read.csv("ENV_pitlatrine.csv",row.names=1,check.names=FALSE) #Just a check to ensure that the samples in meta_table are in the same order as in abund_table meta_table<-meta_table[rownames(abund_table),] #Get grouping information grouping_info<-data.frame(row.names=rownames(abund_table),t(as.data.frame(strsplit(rownames(abund_table),"_")))) # > head(grouping_info) # X1 X2 X3 # T_2_1 T 2 1 # T_2_10 T 2 10 # T_2_12 T 2 12 # T_2_2 T 2 2 # T_2_3 T 2 3 # T_2_6 T 2 6 #Let us group on countries groups<-grouping_info[,1] library(vegan) #Get MDS stats sol<-metaMDS(abund_table,distance = "bray", k = 2, trymax = 50) #We use meta_table$Temp to plot temperature values on the plot. You can select #any other variable #> names(meta_table) #[1] "pH" "Temp" "TS" "VS" "VFA" "CODt" #[7] "CODs" "perCODsbyt" "NH4" "Prot" "Carbo" #Reference:http://oliviarata.wordpress.com/2014/07/17/ordinations-in-ggplot2-v2-ordisurf/ ordi<-ordisurf(sol,meta_table$Temp,plot = FALSE, bs="ds") ordi.grid <- ordi$grid #extracts the ordisurf object str(ordi.grid) #it's a list though - cannot be plotted as is ordi.mite <- expand.grid(x = ordi.grid$x, y = ordi.grid$y) #get x and ys ordi.mite$z <- as.vector(ordi.grid$z) #unravel the matrix for the z scores ordi.mite.na <- data.frame(na.omit(ordi.mite)) #gets rid of the nas NMDS=data.frame(x=sol$point[,1],y=sol$point[,2],Type=groups) p<-ggplot()+ stat_contour(data = ordi.mite.na, aes(x = x, y = y, z = z, colour = ..level..),positon="identity")+ #can change the binwidth depending on how many contours you want geom_point(data=NMDS,aes(x,y,fill=Type),pch=21,size=3,colour=NA) p<-p+scale_colour_continuous(high = "darkgreen", low = "darkolivegreen1") #here we set the high and low of the colour scale. Can delete to go back to the standard blue, or specify others p<-p+labs(colour = "Temperature") #another way to set the labels, in this case, for the colour legend p<-p+theme_bw() #p<-p+theme(legend.key = element_blank(), #removes the box around each legend item # legend.position = "bottom", #legend at the bottom # legend.direction = "horizontal", # legend.box = "horizontal", # legend.box.just = "centre") pdf("NMDS_ordisurf.pdf",width=10,height=10) print(p) dev.off()
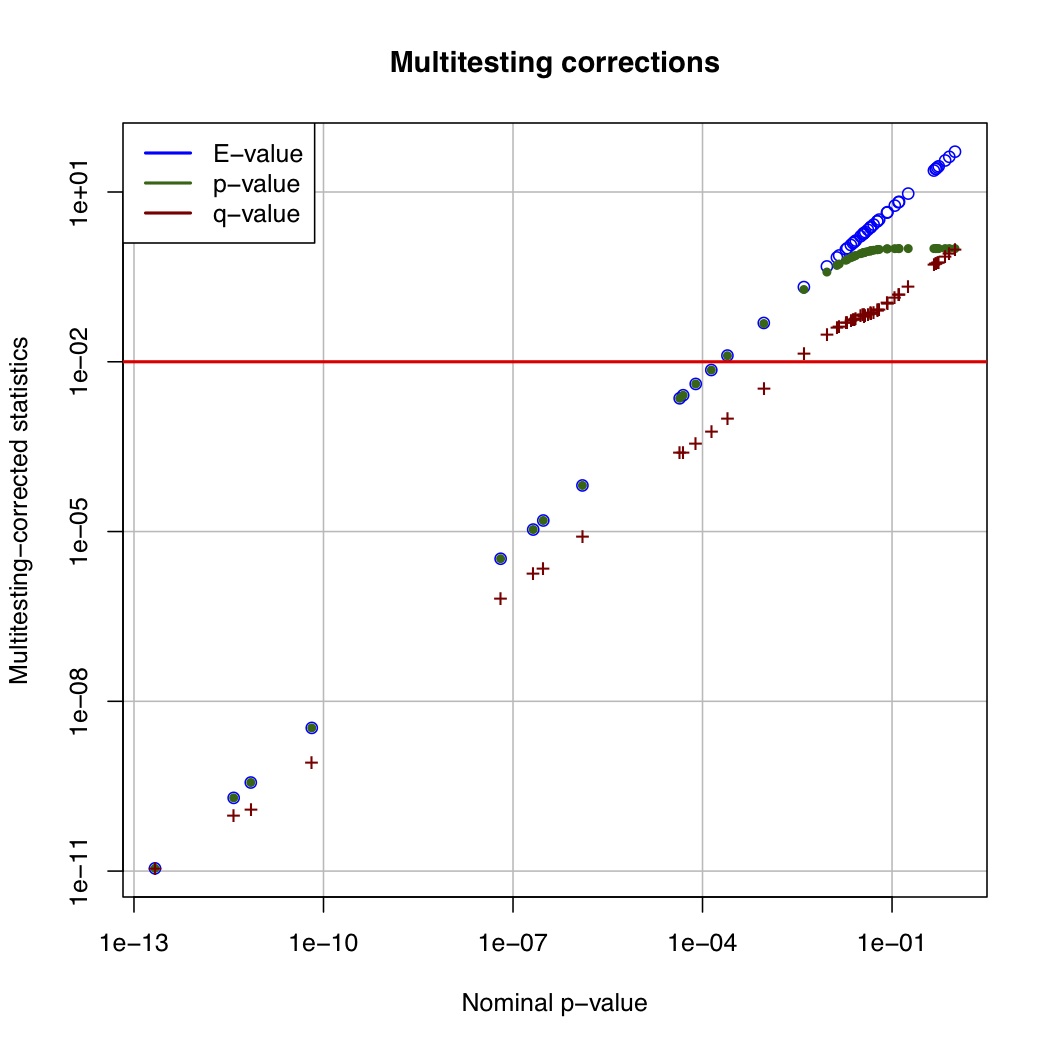
# ============================================================ # Tutorial on finding significant taxa and then plotting them using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= library(reshape) library(ggplot2) #Load the abundance table abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) #Get grouping information grouping_info<-data.frame(row.names=rownames(abund_table),t(as.data.frame(strsplit(rownames(abund_table),"_")))) # > head(grouping_info) # X1 X2 X3 # T_2_1 T 2 1 # T_2_10 T 2 10 # T_2_12 T 2 12 # T_2_2 T 2 2 # T_2_3 T 2 3 # T_2_6 T 2 6 #Use countries as grouping information groups<-as.factor(grouping_info[,1]) #Apply normalisation (either use relative or log-relative transformation) #data<-abund_table/rowSums(abund_table) data<-log((abund_table+1)/(rowSums(abund_table)+dim(abund_table)[2])) data<-as.data.frame(data) #Reference: http://www.bigre.ulb.ac.be/courses/statistics_bioinformatics/practicals/microarrays_berry_2010/berry_feature_selection.html kruskal.wallis.alpha=0.01 kruskal.wallis.table <- data.frame() for (i in 1:dim(data)[2]) { ks.test <- kruskal.test(data[,i], g=groups) # Store the result in the data frame kruskal.wallis.table <- rbind(kruskal.wallis.table, data.frame(id=names(data)[i], p.value=ks.test$p.value )) # Report number of values tested cat(paste("Kruskal-Wallis test for ",names(data)[i]," ", i, "/", dim(data)[2], "; p-value=", ks.test$p.value,"\n", sep="")) } kruskal.wallis.table$E.value <- kruskal.wallis.table$p.value * dim(kruskal.wallis.table)[1] kruskal.wallis.table$FWER <- pbinom(q=0, p=kruskal.wallis.table$p.value, size=dim(kruskal.wallis.table)[1], lower.tail=FALSE) kruskal.wallis.table <- kruskal.wallis.table[order(kruskal.wallis.table$p.value, decreasing=FALSE), ] kruskal.wallis.table$q.value.factor <- dim(kruskal.wallis.table)[1] / 1:dim(kruskal.wallis.table)[1] kruskal.wallis.table$q.value <- kruskal.wallis.table$p.value * kruskal.wallis.table$q.value.factor pdf("KW_correction.pdf") plot(kruskal.wallis.table$p.value, kruskal.wallis.table$E.value, main='Multitesting corrections', xlab='Nominal p-value', ylab='Multitesting-corrected statistics', log='xy', col='blue', panel.first=grid(col='#BBBBBB',lty='solid')) lines(kruskal.wallis.table$p.value, kruskal.wallis.table$FWER, pch=20,col='darkgreen', type='p' ) lines(kruskal.wallis.table$p.value, kruskal.wallis.table$q.value, pch='+',col='darkred', type='p' ) abline(h=kruskal.wallis.alpha, col='red', lwd=2) legend('topleft', legend=c('E-value', 'p-value', 'q-value'), col=c('blue', 'darkgreen','darkred'), lwd=2,bg='white',bty='o') dev.off() last.significant.element <- max(which(kruskal.wallis.table$q.value <= kruskal.wallis.alpha)) selected <- 1:last.significant.element diff.cat.factor <- kruskal.wallis.table$id[selected] diff.cat <- as.vector(diff.cat.factor) print(kruskal.wallis.table[selected,]) #Now we plot taxa significantly different between the categories df<-NULL for(i in diff.cat){ tmp<-data.frame(data[,i],groups,rep(paste(i," q = ",round(kruskal.wallis.table[kruskal.wallis.table$id==i,"q.value"],5),sep=""),dim(data)[1])) if(is.null(df)){df<-tmp} else { df<-rbind(df,tmp)} } colnames(df)<-c("Value","Type","Taxa") p<-ggplot(df,aes(Type,Value,colour=Type))+ylab("Log-relative normalised") p<-p+geom_boxplot()+geom_jitter()+theme_bw()+ facet_wrap( ~ Taxa , scales="free", ncol=3) p<-p+theme(axis.text.x=element_text(angle=90,hjust=1,vjust=0.5)) pdf("KW_significant.pdf",width=10,height=14) print(p) dev.off()
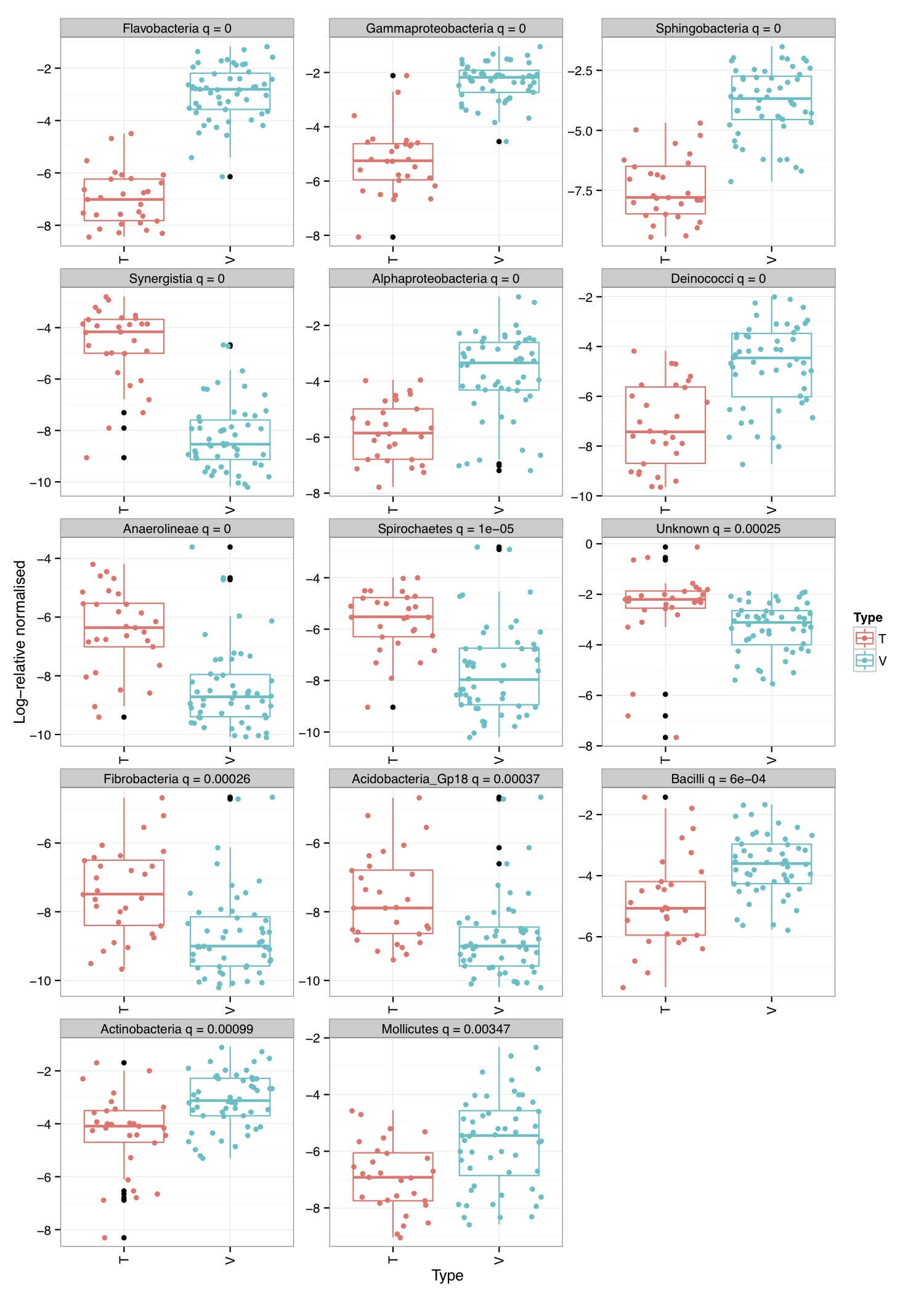
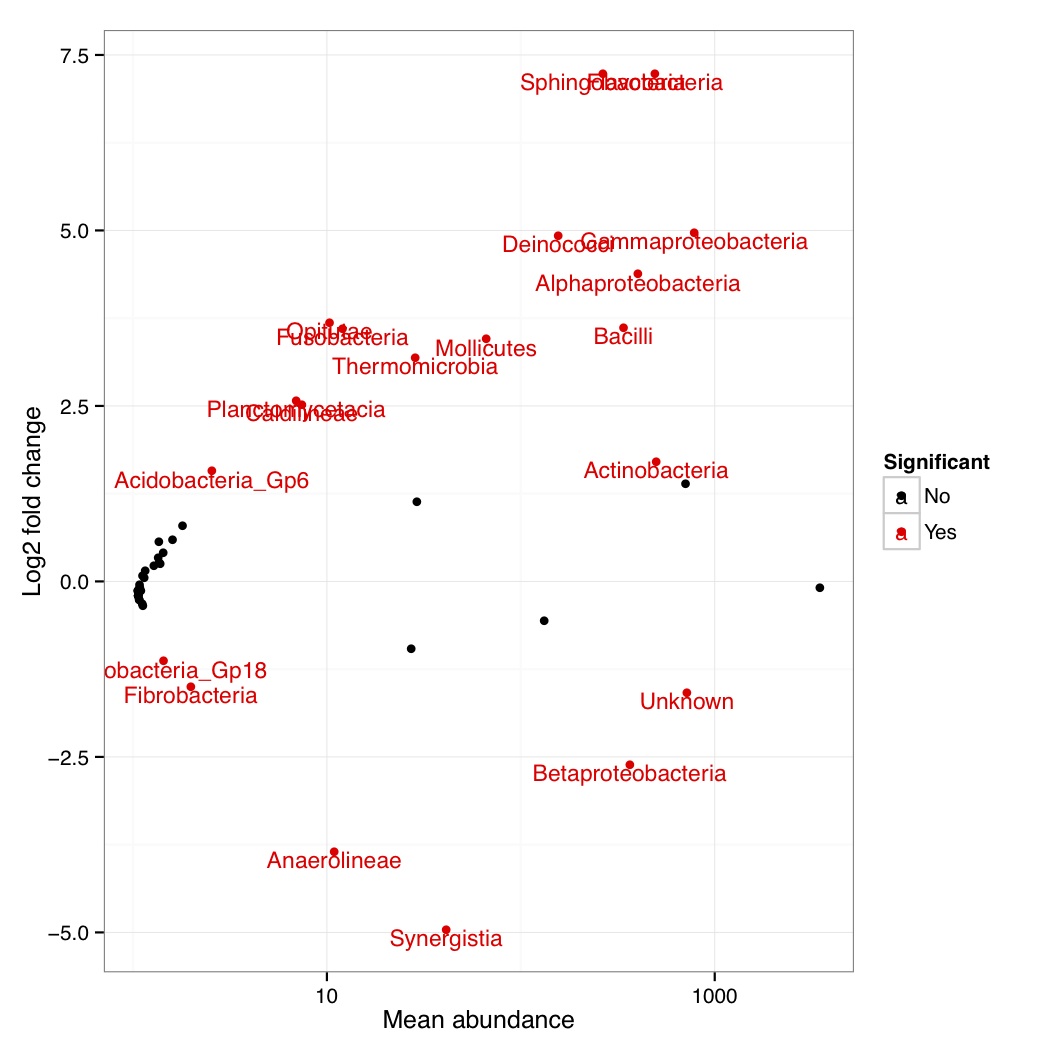
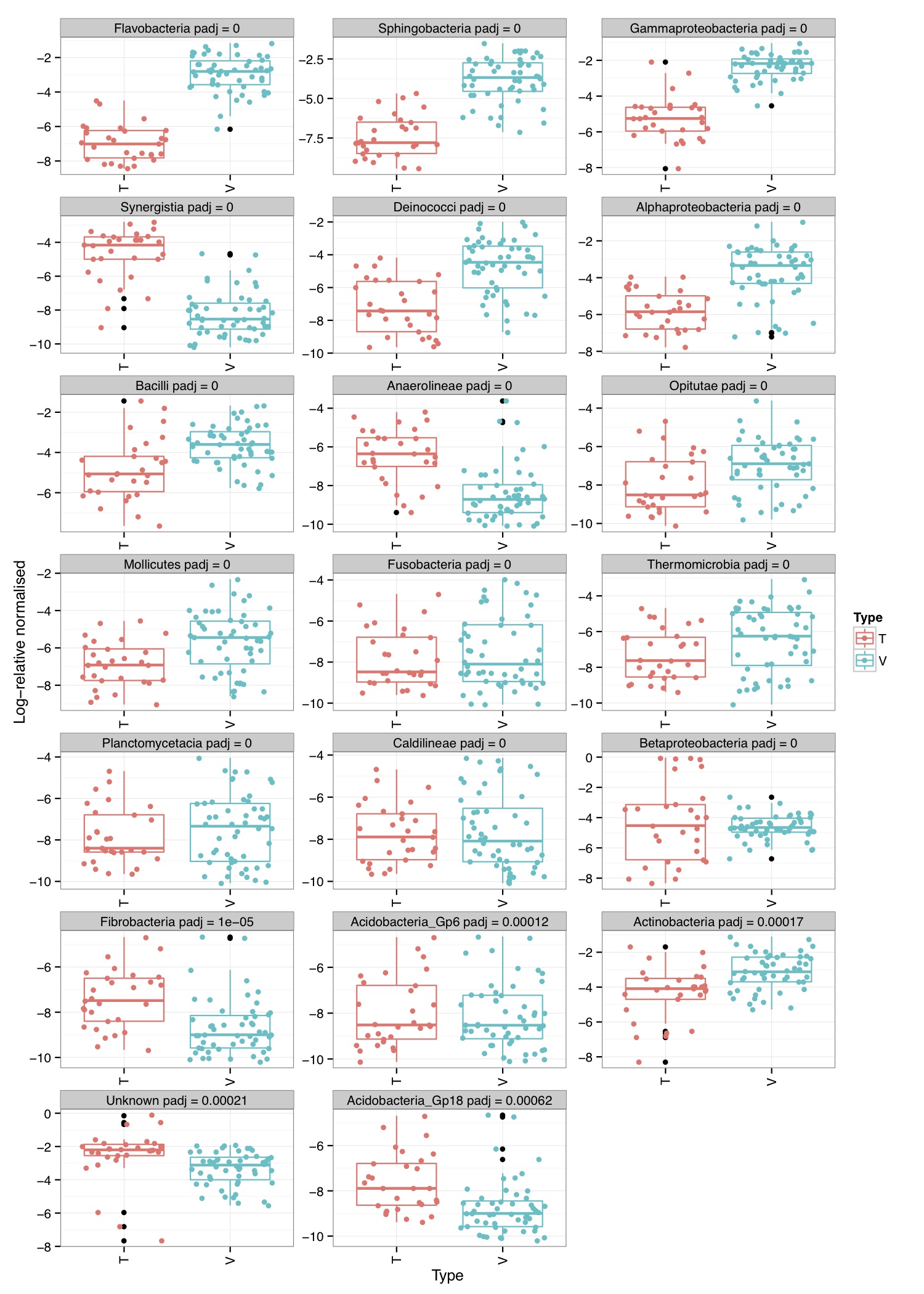
# ============================================================ # Tutorial on finding significant taxa and then plotting them using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= library(ggplot2) library(DESeq2) #Load the abundance table abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) #Get grouping information grouping_info<-data.frame(row.names=rownames(abund_table),t(as.data.frame(strsplit(rownames(abund_table),"_")))) # > head(grouping_info) # X1 X2 X3 # T_2_1 T 2 1 # T_2_10 T 2 10 # T_2_12 T 2 12 # T_2_2 T 2 2 # T_2_3 T 2 3 # T_2_6 T 2 6 #We will convert our table to DESeqDataSet object countData = round(as(abund_table, "matrix"), digits = 0) # We will add 1 to the countData otherwise DESeq will fail with the error: # estimating size factors # Error in estimateSizeFactorsForMatrix(counts(object), locfunc = locfunc, : # every gene contains at least one zero, cannot compute log geometric means countData<-(t(countData+1)) dds <- DESeqDataSetFromMatrix(countData, grouping_info, as.formula(~ X1)) #Reference:https://github.com/MadsAlbertsen/ampvis/blob/master/R/amp_test_species.R #Differential expression analysis based on the Negative Binomial (a.k.a. Gamma-Poisson) distribution #Some reason this doesn't work: data_deseq_test = DESeq(dds, test="wald", fitType="parametric") data_deseq_test = DESeq(dds) ## Extract the results res = results(data_deseq_test, cooksCutoff = FALSE) res_tax = cbind(as.data.frame(res), as.matrix(countData[rownames(res), ]), OTU = rownames(res)) sig = 0.001 fold = 0 plot.point.size = 2 label=T tax.display = NULL tax.aggregate = "OTU" res_tax_sig = subset(res_tax, padj < sig & fold < abs(log2FoldChange)) res_tax_sig <- res_tax_sig[order(res_tax_sig$padj),] ## Plot the data ### MA plot res_tax$Significant <- ifelse(rownames(res_tax) %in% rownames(res_tax_sig) , "Yes", "No") res_tax$Significant[is.na(res_tax$Significant)] <- "No" p1 <- ggplot(data = res_tax, aes(x = baseMean, y = log2FoldChange, color = Significant)) + geom_point(size = plot.point.size) + scale_x_log10() + scale_color_manual(values=c("black", "red")) + labs(x = "Mean abundance", y = "Log2 fold change")+theme_bw() if(label == T){ if (!is.null(tax.display)){ rlab <- data.frame(res_tax, Display = apply(res_tax[,c(tax.display, tax.aggregate)], 1, paste, collapse="; ")) } else { rlab <- data.frame(res_tax, Display = res_tax[,tax.aggregate]) } p1 <- p1 + geom_text(data = subset(rlab, Significant == "Yes"), aes(label = Display), size = 4, vjust = 1) } pdf("NB_MA.pdf") print(p1) dev.off() res_tax_sig_abund = cbind(as.data.frame(countData[rownames(res_tax_sig), ]), OTU = rownames(res_tax_sig), padj = res_tax[rownames(res_tax_sig),"padj"]) #Apply normalisation (either use relative or log-relative transformation) #data<-abund_table/rowSums(abund_table) data<-log((abund_table+1)/(rowSums(abund_table)+dim(abund_table)[2])) data<-as.data.frame(data) #Now we plot taxa significantly different between the categories df<-NULL for(i in res_tax[rownames(res_tax_sig),"OTU"]){ tmp<-data.frame(data[,i],groups,rep(paste(i," padj = ",round(res_tax[i,"padj"],5),sep=""),dim(data)[1])) if(is.null(df)){df<-tmp} else { df<-rbind(df,tmp)} } colnames(df)<-c("Value","Type","Taxa") p<-ggplot(df,aes(Type,Value,colour=Type))+ylab("Log-relative normalised") p<-p+geom_boxplot()+geom_jitter()+theme_bw()+ facet_wrap( ~ Taxa , scales="free", ncol=3) p<-p+theme(axis.text.x=element_text(angle=90,hjust=1,vjust=0.5)) pdf("NB_significant.pdf",width=10,height=14) print(p) dev.off()
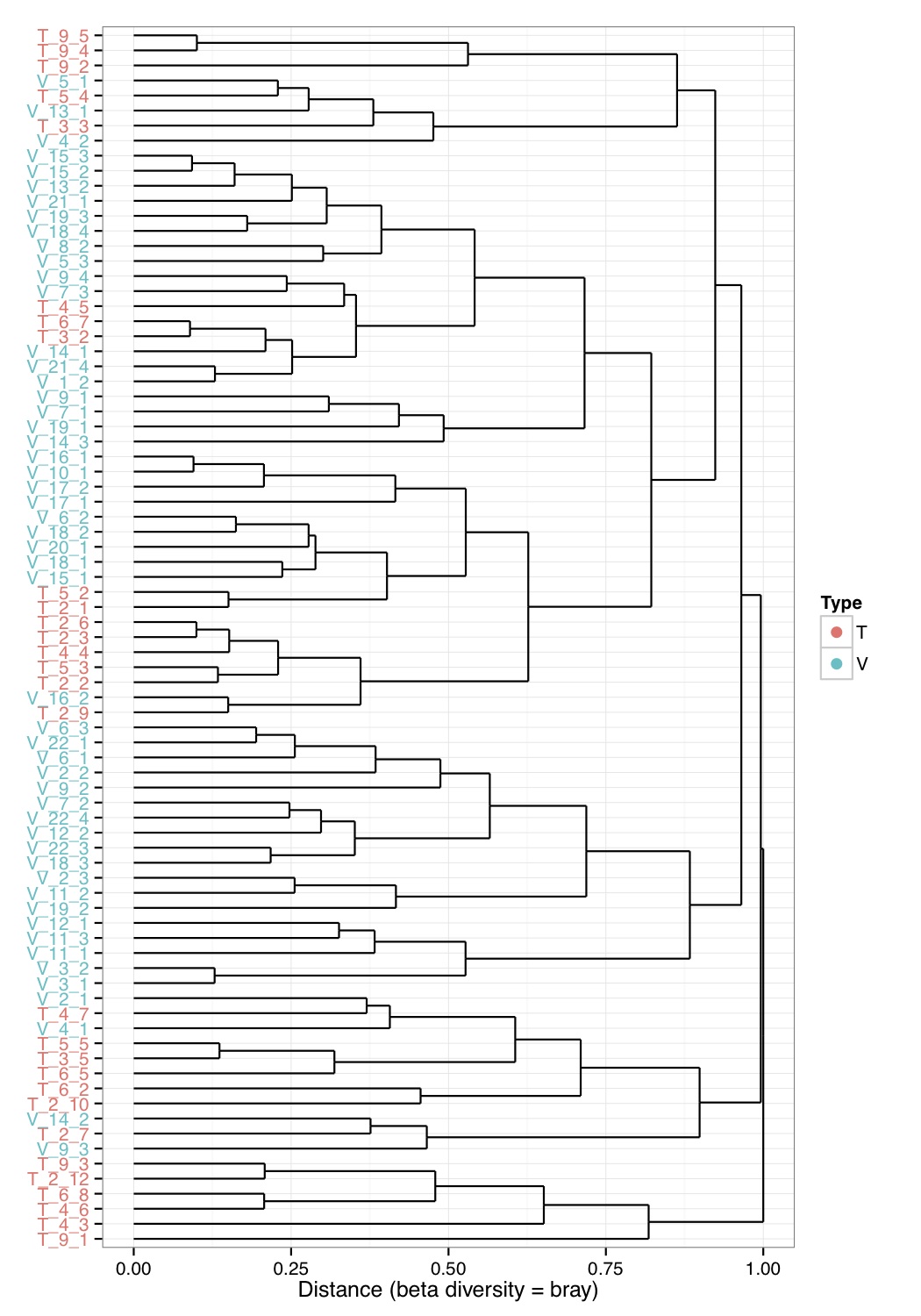
# ============================================================ # Tutorial on hierarchical clustering and plotting samples using ggplot2 # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= library(ggplot2) library(ggdendro) #Load the abundance table abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) #Get grouping information grouping_info<-data.frame(row.names=rownames(abund_table),t(as.data.frame(strsplit(rownames(abund_table),"_")))) # > head(grouping_info) # X1 X2 X3 # T_2_1 T 2 1 # T_2_10 T 2 10 # T_2_12 T 2 12 # T_2_2 T 2 2 # T_2_3 T 2 3 # T_2_6 T 2 6 betad<-vegdist(abund_table,method="bray") # Use Adonis to test for overall differences res_adonis <- adonis(betad ~ X1, grouping_info) #Cluster the samples hc <- hclust(betad) #We will color the labels according to countries(group_info[,1]) hc_d <- dendro_data(as.dendrogram(hc)) hc_d$labels$Type<-grouping_info[as.character(hc_d$labels$label),1] #Coloring function gg_color_hue<-function(n){ hues=seq(15,375,length=n+1) hcl(h=hues,l=65,c=100)[1:n] } cols=gg_color_hue(length(unique(hc_d$labels$Type))) hc_d$labels$color=cols[hc_d$labels$Type] ## Plot clusters p1 <- ggplot(data = segment(hc_d)) + geom_segment(aes(x=x, y=y, xend=xend, yend=yend)) + coord_flip() + scale_x_discrete(labels=label(hc_d)$label) + ylab("Distance (beta diversity = bray)") + theme_bw()+ theme(axis.text.y = element_text(color = hc_d$labels$color), axis.title.y = element_blank()) p1 <- p1 + geom_point(data=hc_d$label, aes(x = x, y = y, color = Type), inherit.aes =F, alpha = 0) p1 <- p1 + guides(colour = guide_legend(override.aes = list(size=3, alpha = 1)))+ scale_color_manual(values = cols) pdf("Cluster.pdf",height=10) print(p1) dev.off()
# ============================================================ # Tutorial on normalisation of abundance tables based on Susan Holmes's paper # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= library(phyloseq) library(DESeq) library(edgeR) #===Normalization functions===# #Reference: http://www.ploscompbiol.org/article/info%3Adoi%2F10.1371%2Fjournal.pcbi.1003531 edgeRnorm = function(physeq, ...){ # Enforce orientation. if( !taxa_are_rows(physeq) ){ physeq <- t(physeq) } x = as(otu_table(physeq), "matrix") # See if adding a single observation, 1, # everywhere (so not zeros) prevents errors # without needing to borrow and modify # calcNormFactors (and its dependent functions) # It did. This fixed all problems. # Can the 1 be reduced to something smaller and still work? x = x + 1 # Now turn into a DGEList y = edgeR::DGEList(counts=x, remove.zeros=TRUE) # Perform edgeR-encoded normalization, using the specified method (...) z = edgeR::calcNormFactors(y, ...) # A check that we didn't divide by zero inside `calcNormFactors` if( !all(is.finite(z$samples$norm.factors)) ){ stop("Something wrong with edgeR::calcNormFactors on this data, non-finite $norm.factors") } return(z) } deseq_varstab = function(physeq, sampleConditions=rep("A", nsamples(physeq)), ...){ # Enforce orientation. if( !taxa_are_rows(physeq) ){ physeq <- t(physeq) } x = as(otu_table(physeq), "matrix") # The same tweak as for edgeR to avoid NaN problems # that cause the workflow to stall/crash. x = x + 1 cds = newCountDataSet(x, sampleConditions) # First estimate library size factors cds = estimateSizeFactors(cds) # Variance estimation, passing along additional options cds = estimateDispersions(cds, ...) # Determine which column(s) have the dispersion estimates dispcol = grep("disp\\_", colnames(fData(cds))) # Enforce that there are no infinite values in the dispersion estimates if( any(!is.finite(fData(cds)[, dispcol])) ){ fData(cds)[which(!is.finite(fData(cds)[, dispcol])), dispcol] <- 0.0 } vsmat = exprs(varianceStabilizingTransformation(cds)) otu_table(physeq) <- otu_table(vsmat, taxa_are_rows=TRUE) return(physeq) } proportion = function(physeq){ # Normalize total sequences represented normf = function(x, tot=max(sample_sums(physeq))){ tot*x/sum(x) } physeq = transform_sample_counts(physeq, normf) # Scale by dividing each variable by its standard deviation. #physeq = transform_sample_counts(physeq, function(x) x/sd(x)) # Center by subtracting the median #physeq = transform_sample_counts(physeq, function(x) (x-median(x))) return(physeq) } randomsubsample = function(physeq, smalltrim=0.15, replace=TRUE){ # Set the minimum value as the smallest library quantile, n`smalltrim` samplemin = sort(sample_sums(physeq))[-(1:floor(smalltrim*nsamples(physeq)))][1] physeqr = rarefy_even_depth(physeq, samplemin, rngseed=TRUE, replace=replace, trimOTUs=TRUE) return(physeqr) } #/===Normalization functions===# abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) #We first need to convert table in phyloseq format abund_table_phyloseq=phyloseq(otu_table(abund_table, taxa_are_rows = FALSE)) #Additionally we give it grouping data grouping_info<-data.frame(row.names=rownames(abund_table),t(as.data.frame(strsplit(rownames(abund_table),"_")))) # > head(grouping_info) # X1 X2 X3 # T_2_1 T 2 1 # T_2_10 T 2 10 # T_2_12 T 2 12 # T_2_2 T 2 2 # T_2_3 T 2 3 # T_2_6 T 2 6 sampleData=sample_data(grouping_info) abund_table_phyloseq=merge_phyloseq(abund_table_phyloseq, sampleData) #> abund_table_phyloseq #phyloseq-class experiment-level object #otu_table() OTU Table: [ 52 taxa and 81 samples ] #sample_data() Sample Data: [ 81 samples by 3 sample variables ] #Now apply normalisation methods: #Proportions a<-proportion(abund_table_phyloseq) abund_table_N_proportions<-as.data.frame(otu_table(a)) #Rarefy b<-randomsubsample(abund_table_phyloseq, smalltrim=0.15, replace=FALSE) abund_table_phyloseq_N_rarefied<-as.data.frame(t(otu_table(b))) # method="TMM" is the weighted trimmed mean of M-values # (to the reference) proposed by Robinson and Oshlack (2010), # where the weights are from the delta method on Binomial data. c<-edgeRnorm(abund_table_phyloseq,method="TMM") abund_table_N_edgeRTMM<-as.data.frame(t(c$counts)) # method="RLE" is the scaling factor method proposed by # Anders and Huber (2010). We call it "relative log expression", # as median library is calculated from the geometric mean of all # columns and the median ratio of each sample to the median library # is taken as the scale factor. d<-edgeRnorm(abund_table_phyloseq,method="RLE") abund_table_N_edgeRRLE<-as.data.frame(t(d$counts)) # method="upperquartile" is the upper-quartile normalization method # of Bullard et al (2010), in which the scale factors are calculated # from the 75% quantile of the counts for each library, after removing # transcripts which are zero in all libraries. This idea is generalized # here to allow scaling by any quantile of the distributions. e<-edgeRnorm(abund_table_phyloseq,method="upperquartile") #UQ-logFC method abund_table_N_edgeRupperquartile<-as.data.frame(t(e$counts)) # DESeq variance stabilizing transformations f<-deseq_varstab(abund_table_phyloseq, method="blind", sharingMode="maximum", fitType="local") abund_table_N_varstab<-as.data.frame(t(otu_table(f)))
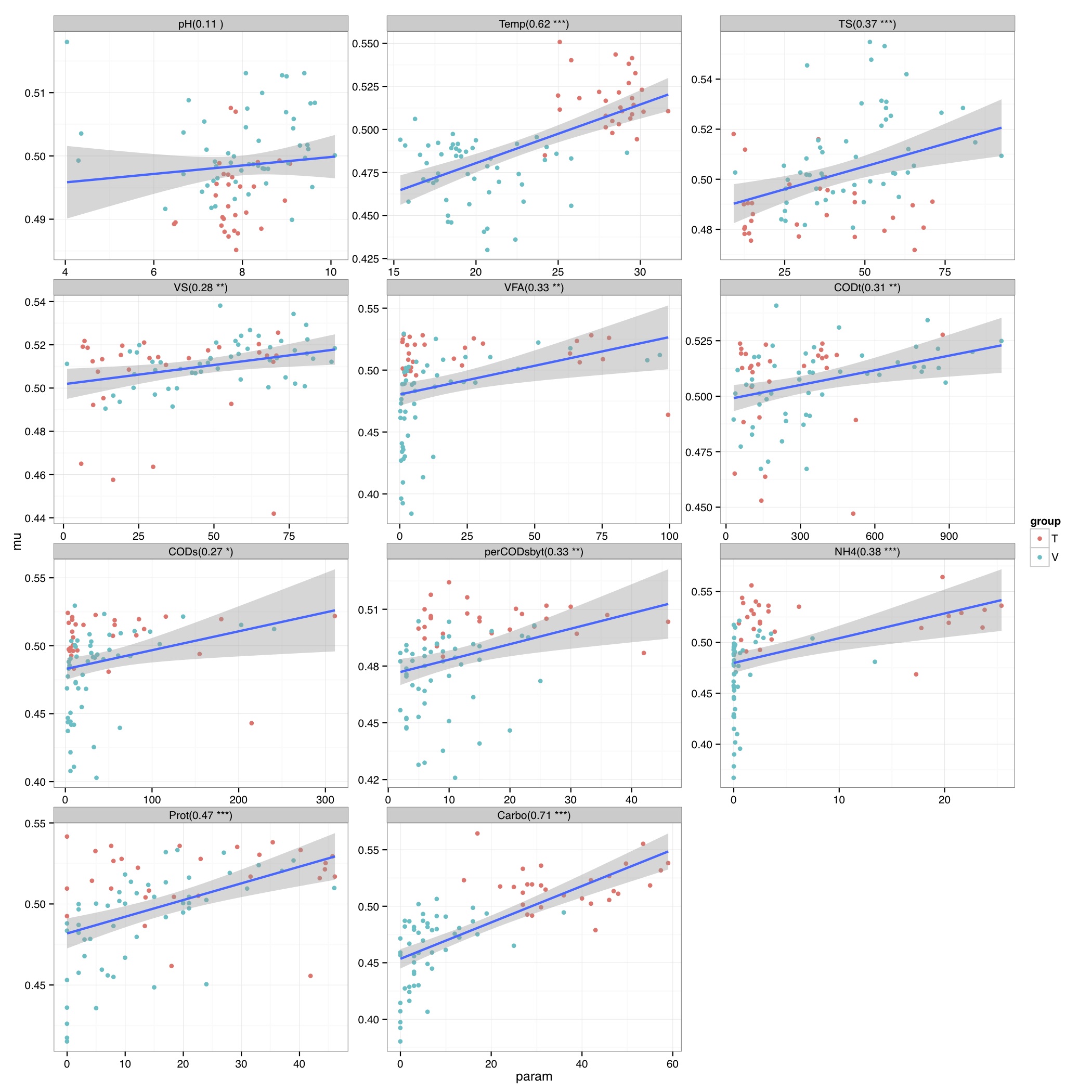
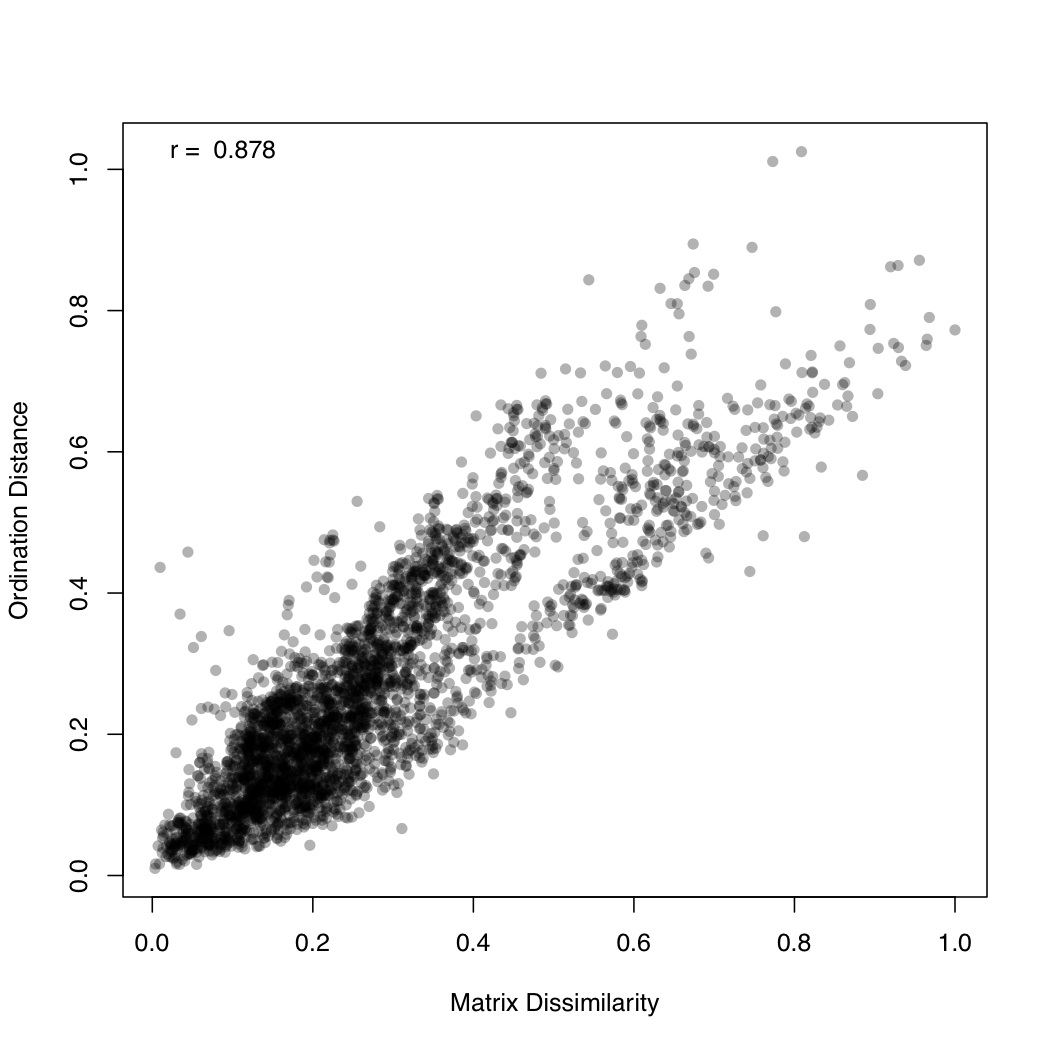
# ============================================================ # Tutorial on fuzzy set ordination # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= library(fso) library(ggplot2) library(vegan) #===dependencies===# #We need a Pvalue formatter formatPvalues <- function(pvalue) { ra<-"" if(pvalue <= 0.1) ra<-"." if(pvalue <= 0.05) ra<-"*" if(pvalue <= 0.01) ra<-"**" if(pvalue <= 0.001) ra<-"***" return(ra) } #Reference: http://www.nku.edu/~boycer/fso/ sim.binary <- function (df, method = NULL, diag=FALSE, upper=FALSE){ df <- as.matrix(df) a <- df %*% t(df) b <- df %*% (1 - t(df)) c <- (1 - df) %*% t(df) d <- ncol(df) - a - b - c #1=Jaccard, 2=Baroni-Urbani & Buser, 3=Kulczynski, 4=Ochiai, 5=Sorensen if (method == 1) { sim <- a/(a + b + c) } else if (method == 2) { sim <- (a + sqrt(a*d))/(a + b + c + sqrt(a*d)) } else if (method == 3) { sim <- 0.5* (a/(a + b) + a/(a + c)) } else if (method == 4) { sim <- a/sqrt((a + b) * (a + c)) } else if (method == 5) { sim <- 2 * a/(2 * a + b + c) } sim2 <- sim[row(sim) > col(sim)] class(sim2) <- "dist" attr(sim2, "Labels") <- dimnames(df)[[1]] attr(sim2, "Diag") <- diag attr(sim2, "Upper") <- upper attr(sim2, "Size") <- nrow(df) attr(sim2, "call") <- match.call() return(sim2) } sim.abund <- function (df, method = NULL, diag = FALSE, upper = FALSE) { METHODS <- c("Baroni-Urbani & Buser", "Horn", "Yule (Modified)") if (!inherits(df, "data.frame")) stop("df is not a data.frame") if (any(df < 0)) stop("non negative value expected in df") if (is.null(method)) { cat("1 = Baroni-Urbani & Buser\n") cat("2 = Horn\n") cat("3 = Yule\n") cat("Select an integer (1-3): ") method <- as.integer(readLines(n = 1)) } df <- as.matrix(df) sites <- nrow(df) species <- ncol(df) sim <- array(0, c(as.integer(sites),as.integer(sites))) spmax <- apply(df,2,max) if (method == 1) { #compute similarities (Baroni-Urbani & Buser) for (x in 1:sites) { for (y in 1:sites) { h1 <- 0 h2 <- 0 h3 <- 0 for (i in 1:species) { h1 <- h1 + min(df[x,i],df[y,i]) h2 <- h2 + max(df[x,i],df[y,i]) h3 <- h3 + spmax[i] - max(df[x,i],df[y,i]) } numer <- h1 + sqrt(h1*h3) denom <- h2 + sqrt(h1*h3) sim[x,y] <- ifelse(identical(denom,0), 0, numer/denom) } } } else if (method == 2) { #compute similarities (Horn) for (x in 1:sites) { for (y in 1:sites) { h1 <- 0 h2 <- 0 h3 <- 0 for (i in 1:species) { if((df[x,i] + df[y,i]) > 0) h1 <- h1 + (df[x,i] + df[y,i]) * log10(df[x,i] + df[y,i]) if(df[x,i] > 0) h2 <- h2 + df[x,i] * log10(df[x,i]) if(df[y,i] > 0) h3 <- h3 + df[y,i] * log10(df[y,i]) } x.sum <- sum(df[x,]) y.sum <- sum(df[y,]) xy.sum <- x.sum + y.sum if (identical(xy.sum, 0)) (sim[x,y] <- 0) else (sim[x,y] <- (h1 - h2 - h3)/(xy.sum * log10(xy.sum)-x.sum * log10(x.sum) - y.sum * log10(y.sum))) if (sim[x,y] < 1.0e-10) sim[x,y] <- 0 } } } else if (method == 3) { #compute similarities (Yule) for (x in 1:sites) { for (y in 1:sites) { h1 <- 0 h2 <- 0 h3 <- 0 h4 <- 0 for (i in 1:species) { h1 <- h1 + min(df[x,i], df[y,i]) h2 <- h2 + max(df[x,i], df[y,i]) - df[y,i] h3 <- h3 + max(df[x,i], df[y,i]) - df[x,i] h4 <- h4 + spmax[i] - max(df[x,i], df[y,i]) } numer <- sqrt(h1*h4) denom <- sqrt(h1*h4) + sqrt(h2*h3) sim[x,y] <- ifelse(identical(denom,0), 0, numer/denom) } } } else stop("Non convenient method") sim >- t(sim) sim2 <- sim[row(sim) > col(sim)] attr(sim2, "Size") <- sites attr(sim2, "Labels") <- dimnames(df)[[1]] attr(sim2, "Diag") <- diag attr(sim2, "Upper") <- upper attr(sim2, "method") <- METHODS[method] attr(sim2, "call") <- match.call() class(sim2) <- "dist" return(sim2) } st.acr <- function(sim, diag=FALSE, upper = FALSE) { library(vegan) dis <- 1 - sim #use "shortest" or "extended" edis <- as.matrix(stepacross(dis, path = "shortest", toolong = 1)) sim <- 1 - edis amax <- max(sim) amin <- min(sim) sim <- (sim-amin)/(amax-amin) sim2 <- sim[row(sim) > col(sim)] attr(sim2, "Size") <- nrow(sim) attr(sim2, "Labels") <- dimnames(sim)[[1]] attr(sim2, "Diag") <- diag attr(sim2, "Upper") <- upper attr(sim2, "call") <- match.call() class(sim2) <- "dist" return(sim2) } #/===dependencies===# #===generateFSO===# #Example usage: # data<- otu_table #(N X P) # Env<- env_table #(N X Q) # group<-as.character(group_categories) #(N X 1) # method<-2 # 1 = Baroni-Urbani & Buser, 2 = Horn, 3 = Yule # indices<-c(1,2) #Specify column no. of Env table; indices<-NULL will take all columns # generateFSO(data,Env,group,method,indices=NULL,"FSO") generateFSO<-function(data,Env,group,method,indices,filename){ if(is.null(indices)){ indices<-seq(1:dim(Env)[2]) } cat("Calculating dissimilarity matrix\n") sim <- sim.abund(data,method=method) dis.ho <- 1 - sim df<-NULL for(i in names(Env)[indices]){ cat(paste("Processing",i,"\n")) param.fso<-fso(Env[,i],dis.ho,permute=1000) tmp<-data.frame(mu=param.fso$mu,param=param.fso$data, group=as.factor(group),label=rep(paste(i,"(",round(param.fso$r,2)," ",formatPvalues(param.fso$p),")",sep=""),dim(data)[1])) if(is.null(df)){df<-tmp} else {df<-rbind(df,tmp)} } pdf(paste(filename,".pdf",sep=""),height=14,width=14) p <- ggplot(df, aes(param, mu)) + geom_point(aes(colour = group)) +geom_smooth(,method="lm", size=1, se=T) +theme_bw()+ facet_wrap( ~ label , scales="free", ncol=3) print(p) dev.off() whole.fso<-fso(as.formula(paste("~",paste(lapply(indices,function(x) paste("Env[,",x,"]",sep="")),collapse="+"))),dis=dis.ho,data=Env,permute=1000) whole.fso$var<-names(Env)[indices] print(summary(whole.fso)) } #===/generateFSO===# #===generateMFSO===# #Example usage: # data<- otu_table #(N X P) # Env<- env_table #(N X Q) # group<-as.character(group_categories) #(N X 1) # method<-2 # 1 = Baroni-Urbani & Buser, 2 = Horn, 3 = Yule # indices<-c(1,2) #Specify column no. of Env table; indices<-NULL will take all columns # step_across<-TRUE #Step-across correction might improve the ordination # generateMFSO(data,Env,group,method,indices=NULL,step_across=TRUE,"MFSO") generateMFSO<-function(data,Env,group,method,indices,step_across=FALSE,filename){ if(is.null(indices)){ indices<-seq(1:dim(Env)[2]) } cat("Calculating dissimilarity matrix\n") sim <- sim.abund(data,method=method) if(step_across){sim <- st.acr(sim)} dis.ho <- 1 - sim whole.mfso<-mfso(as.formula(paste("~",paste(lapply(indices,function(x) paste("Env[,",x,"]",sep="")),collapse="+"))),dis=dis.ho,data=Env,scaling=2,permute=1000) whole.mfso$var<-names(Env)[indices] names(whole.mfso$data)<-names(Env)[indices] df<-NULL for (i in 1:(ncol(whole.mfso$mu) - 1)) { for (j in (i + 1):ncol(whole.mfso$mu)) { cat(paste("Processing",names(whole.mfso$data)[i]," and ",names(whole.mfso$data)[j],"\n")) tmp<-data.frame(x=whole.mfso$mu[, i],y=whole.mfso$mu[, j], group=as.factor(group),label=rep(paste("x=mu(",names(whole.mfso$data)[i],")",", y=mu(",names(whole.mfso$data)[j],")",sep=""),dim(data)[1])) if(is.null(df)){df<-tmp} else {df<-rbind(df,tmp)} } } pdf(paste(filename,"_mu.pdf",sep=""),height=35,width=14) p <- ggplot(df, aes(x, y)) + geom_point(aes(colour = group)) +geom_smooth(,method="lm", size=1, se=T) +theme_bw()+ facet_wrap( ~ label , scales="free", ncol=4) print(p) dev.off() tmp <- as.numeric(whole.mfso$notmis) dis <- as.matrix(dis.ho)[tmp, tmp] a <- dist(whole.mfso$mu) y <- as.dist(dis) pdf(paste(filename,"_correlation.pdf",sep="")) plot(y, a, ylab = "Ordination Distance", xlab = "Matrix Dissimilarity",pch=16,col=rgb(0, 0, 0, 0.3)) text(min(y), max(a), paste("r = ", format(cor(y, a), digits = 3)), pos = 4) dev.off() print(summary(whole.mfso)) } #===/generateMFSO===# abund_table<-read.csv("SPE_pitlatrine.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) meta_table<-read.csv("ENV_pitlatrine.csv",row.names=1,check.names=FALSE) #Just a check to ensure that the samples in meta_table are in the same order as in abund_table meta_table<-meta_table[rownames(abund_table),] #Get grouping information grouping_info<-data.frame(row.names=rownames(abund_table),t(as.data.frame(strsplit(rownames(abund_table),"_")))) # > head(grouping_info) # X1 X2 X3 # T_2_1 T 2 1 # T_2_10 T 2 10 # T_2_12 T 2 12 # T_2_2 T 2 2 # T_2_3 T 2 3 # T_2_6 T 2 6 generateFSO(as.data.frame(abund_table),meta_table,grouping_info[,1],2,NULL,"FSO") # Fuzzy set statistics # # col variable r p d # 1 11 Carbo 0.7140058 0.001 0.6943693 # 2 2 Temp 0.6202771 0.001 0.7671225 # 3 10 Prot 0.4659610 0.001 0.5658457 # 4 9 NH4 0.3819395 0.001 0.5115750 # 5 3 TS 0.3653211 0.002 0.5182094 # 6 8 perCODsbyt 0.3338634 0.004 0.5630355 # 7 5 VFA 0.3289921 0.004 0.4786655 # 8 6 CODt 0.3126350 0.003 0.8194612 # 9 4 VS 0.2768421 0.006 0.8264804 # 10 7 CODs 0.2717105 0.007 0.5133256 # 11 1 pH 0.1052026 0.153 0.5623866 # # Correlations of fuzzy sets # # pH Temp TS VS VFA CODt # pH 1.0000000 -0.7386860 0.9647511 -0.53378849 -0.9269124 -0.7363250 # Temp -0.7386860 1.0000000 -0.7662551 -0.13447847 0.8173559 0.1627051 # TS 0.9647511 -0.7662551 1.0000000 -0.52324123 -0.9783639 -0.7529399 # VS -0.5337885 -0.1344785 -0.5232412 1.00000000 0.4241976 0.9459625 # VFA -0.9269124 0.8173559 -0.9783639 0.42419761 1.0000000 0.6793673 # CODt -0.7363250 0.1627051 -0.7529399 0.94596254 0.6793673 1.0000000 # CODs -0.9336815 0.7106207 -0.9818299 0.57275257 0.9839748 0.7936315 # perCODsbyt -0.8789800 0.9163897 -0.9321846 0.22357388 0.9708713 0.5029946 # NH4 -0.9456583 0.8863344 -0.9595074 0.32493987 0.9686050 0.5826021 # Prot -0.9656359 0.6903348 -0.9890375 0.61667884 0.9636521 0.8233212 # Carbo -0.8189456 0.9819195 -0.8547344 0.02029398 0.9018883 0.3181941 # CODs perCODsbyt NH4 Prot Carbo # pH -0.9336815 -0.8789800 -0.9456583 -0.9656359 -0.81894558 # Temp 0.7106207 0.9163897 0.8863344 0.6903348 0.98191952 # TS -0.9818299 -0.9321846 -0.9595074 -0.9890375 -0.85473437 # VS 0.5727526 0.2235739 0.3249399 0.6166788 0.02029398 # VFA 0.9839748 0.9708713 0.9686050 0.9636521 0.90188831 # CODt 0.7936315 0.5029946 0.5826021 0.8233212 0.31819413 # CODs 1.0000000 0.9212524 0.9342960 0.9858916 0.81594415 # perCODsbyt 0.9212524 1.0000000 0.9704735 0.8894673 0.96957738 # NH4 0.9342960 0.9704735 1.0000000 0.9362088 0.94362521 # Prot 0.9858916 0.8894673 0.9362088 1.0000000 0.79378163 # Carbo 0.8159442 0.9695774 0.9436252 0.7937816 1.00000000 generateMFSO(as.data.frame(abund_table),meta_table,grouping_info[,1],2,indices=NULL,step_across=TRUE,"MFSO") # Statistics for multidimensional fuzzy set ordination # # variable cumulative_r increment p-value gamma # 1 pH 0.5747713 5.747713e-01 0.847 1.000000000 # 2 Temp 0.8611518 2.863805e-01 0.196 0.312136124 # 3 TS 0.8664168 5.265049e-03 0.725 0.027349087 # 4 VS 0.8696778 3.261003e-03 0.775 0.124736229 # 5 VFA 0.8715679 1.890017e-03 0.773 0.012001049 # 6 CODt 0.8757048 4.136928e-03 0.591 0.050717093 # 7 CODs 0.8756964 -8.426964e-06 0.718 0.001398279 # 8 perCODsbyt 0.8761959 4.995468e-04 0.732 0.003167491 # 9 NH4 0.8767607 5.648345e-04 0.515 0.008190486 # 10 Prot 0.8774340 6.732944e-04 0.645 0.004340200 # 11 Carbo 0.8775444 1.104193e-04 0.707 0.001035478
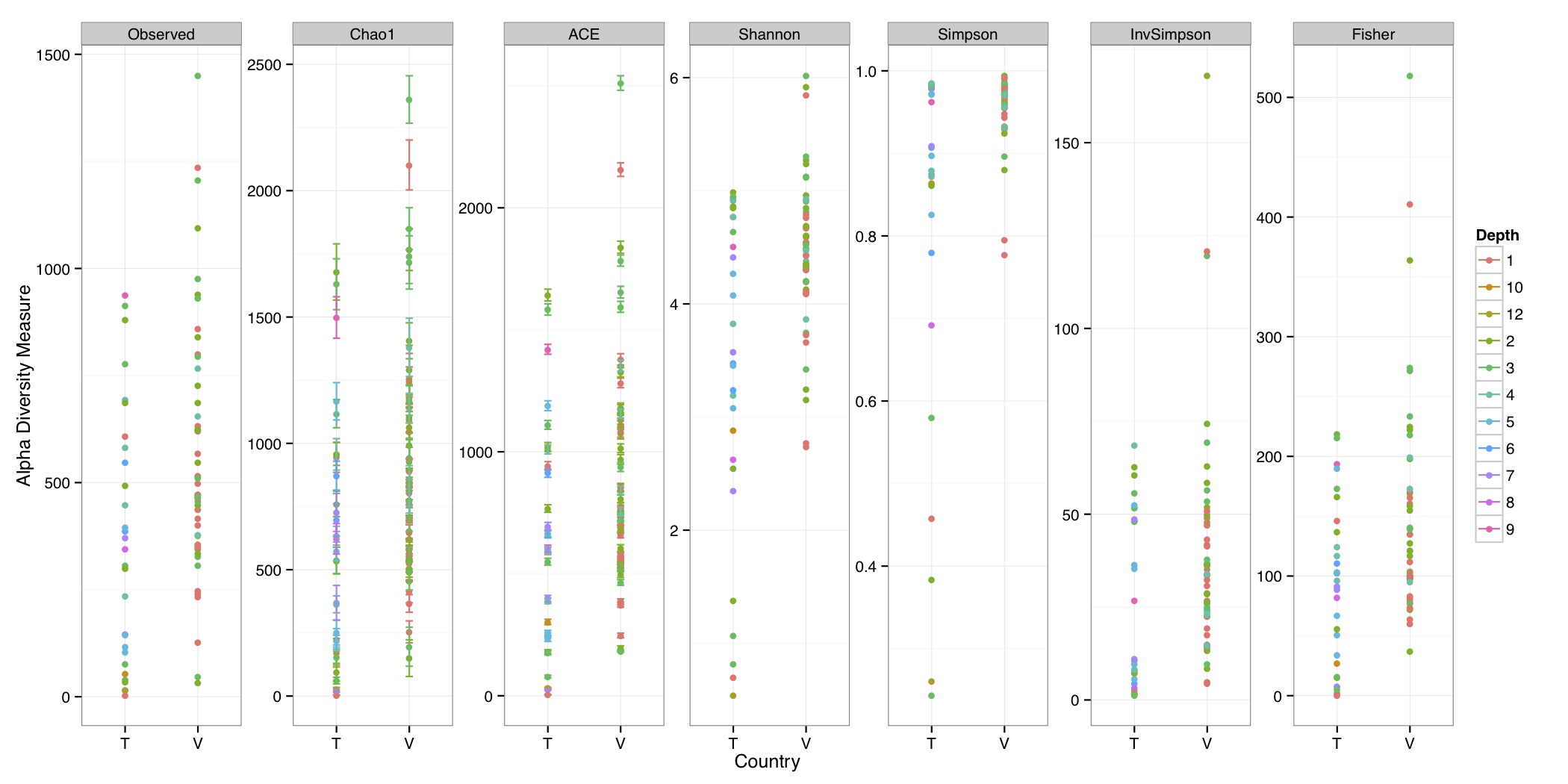
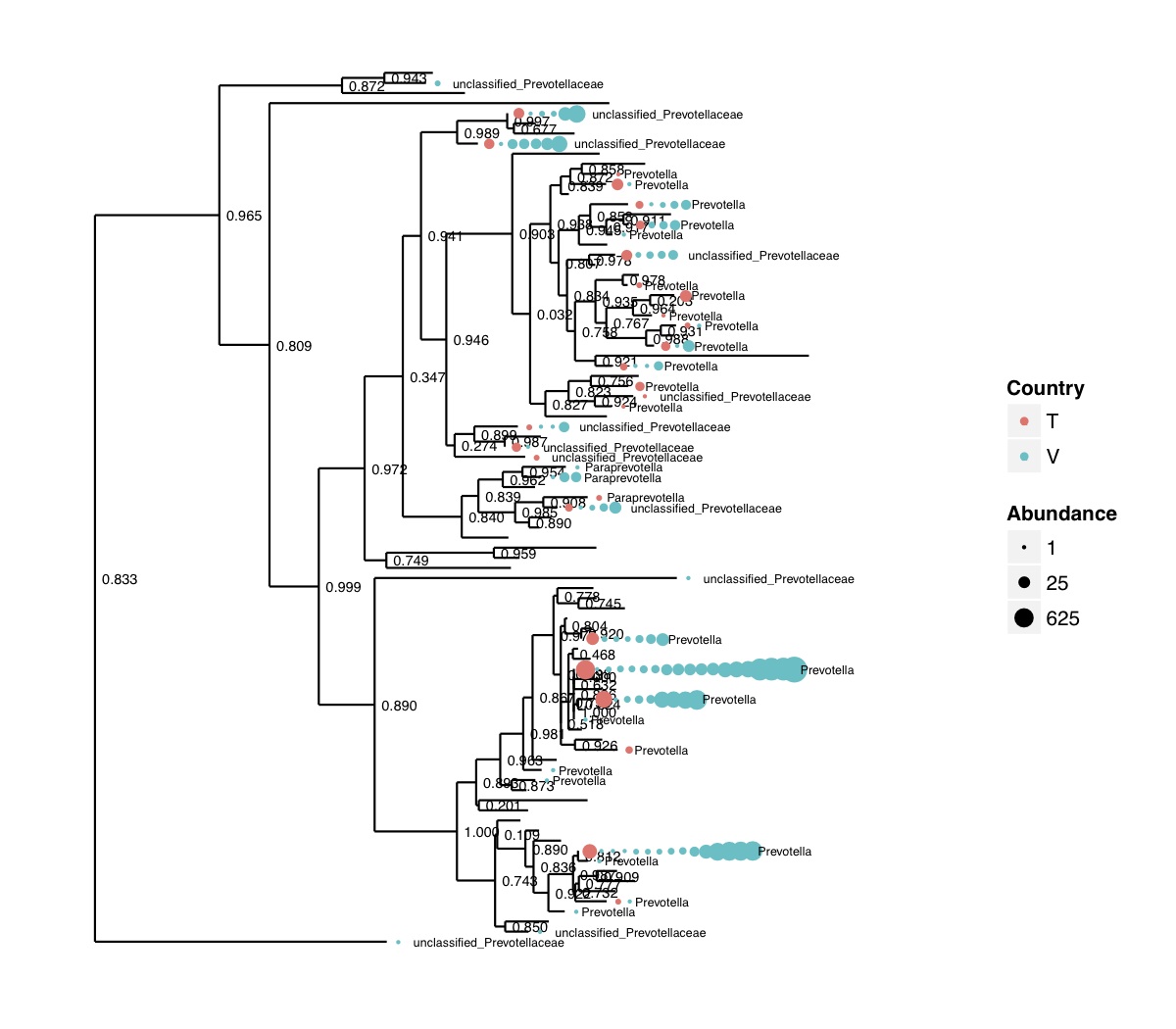
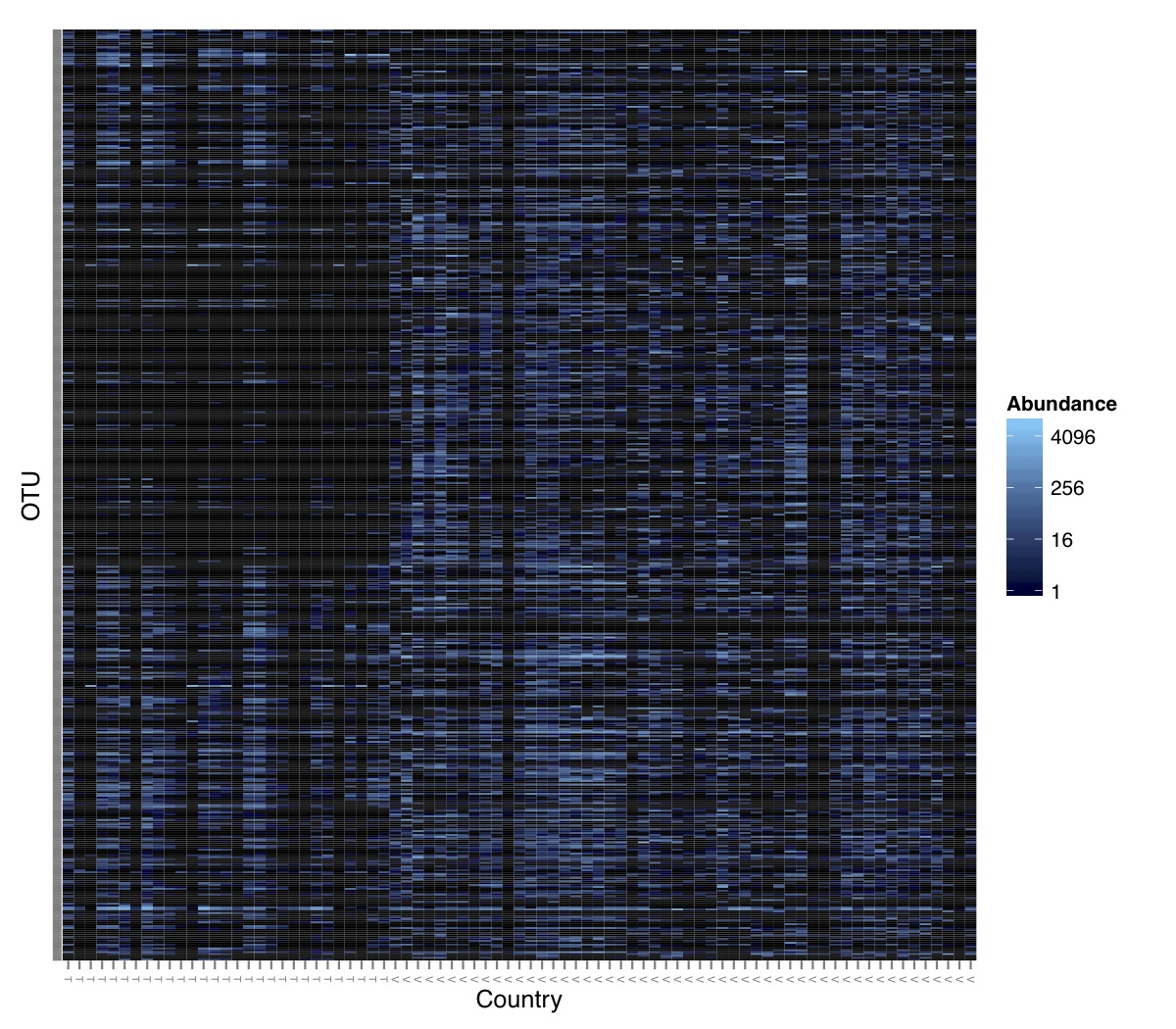
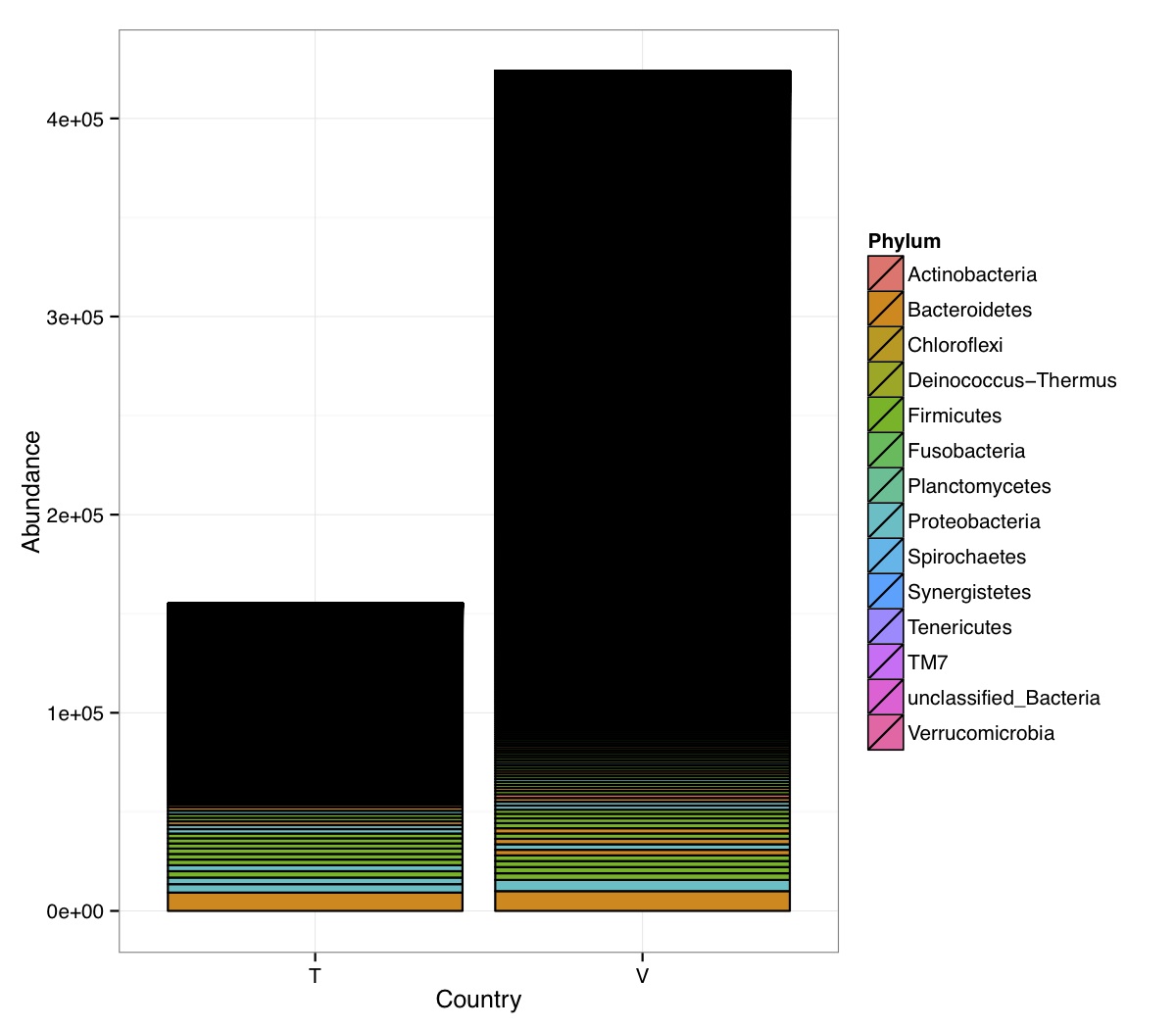
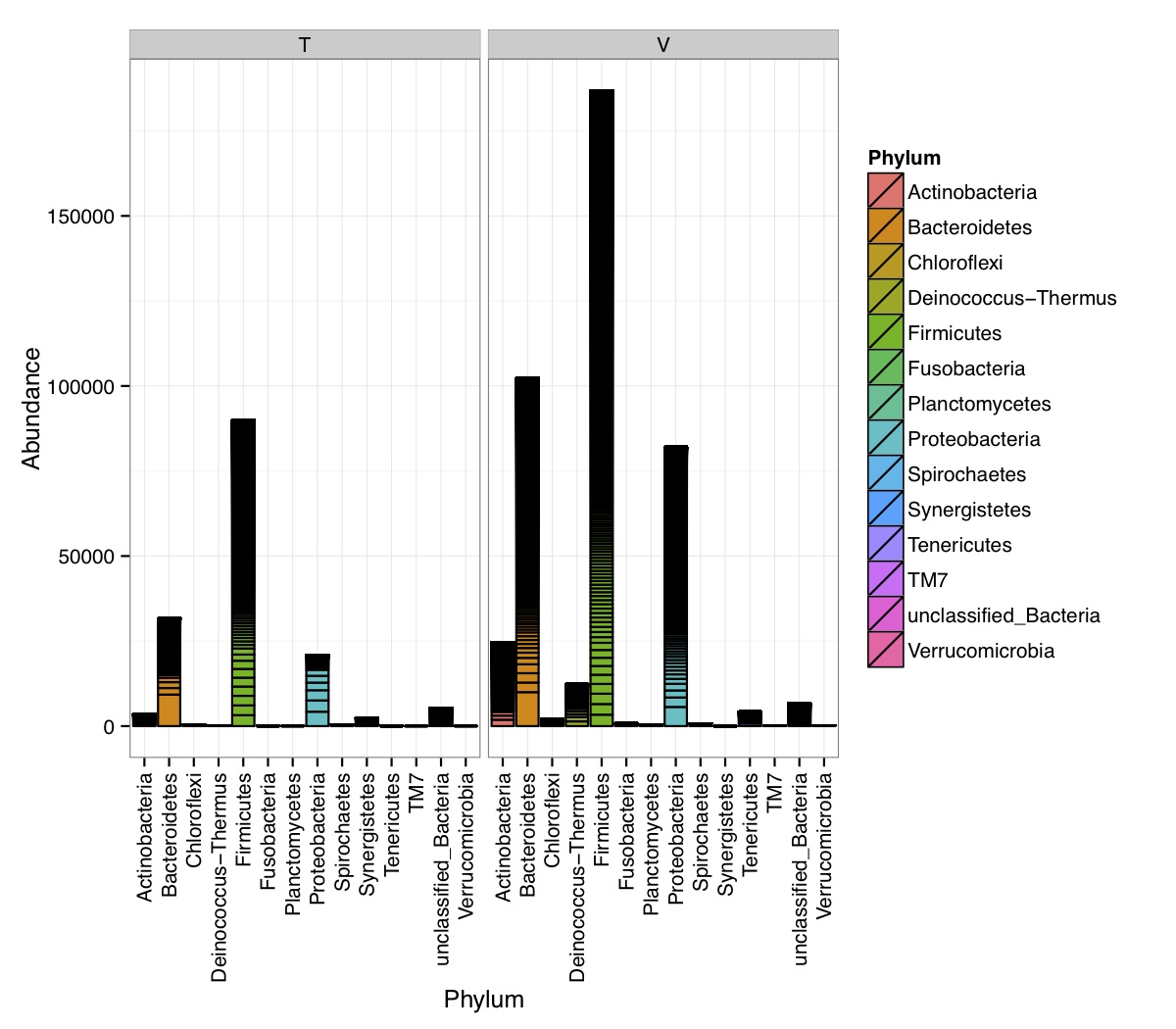
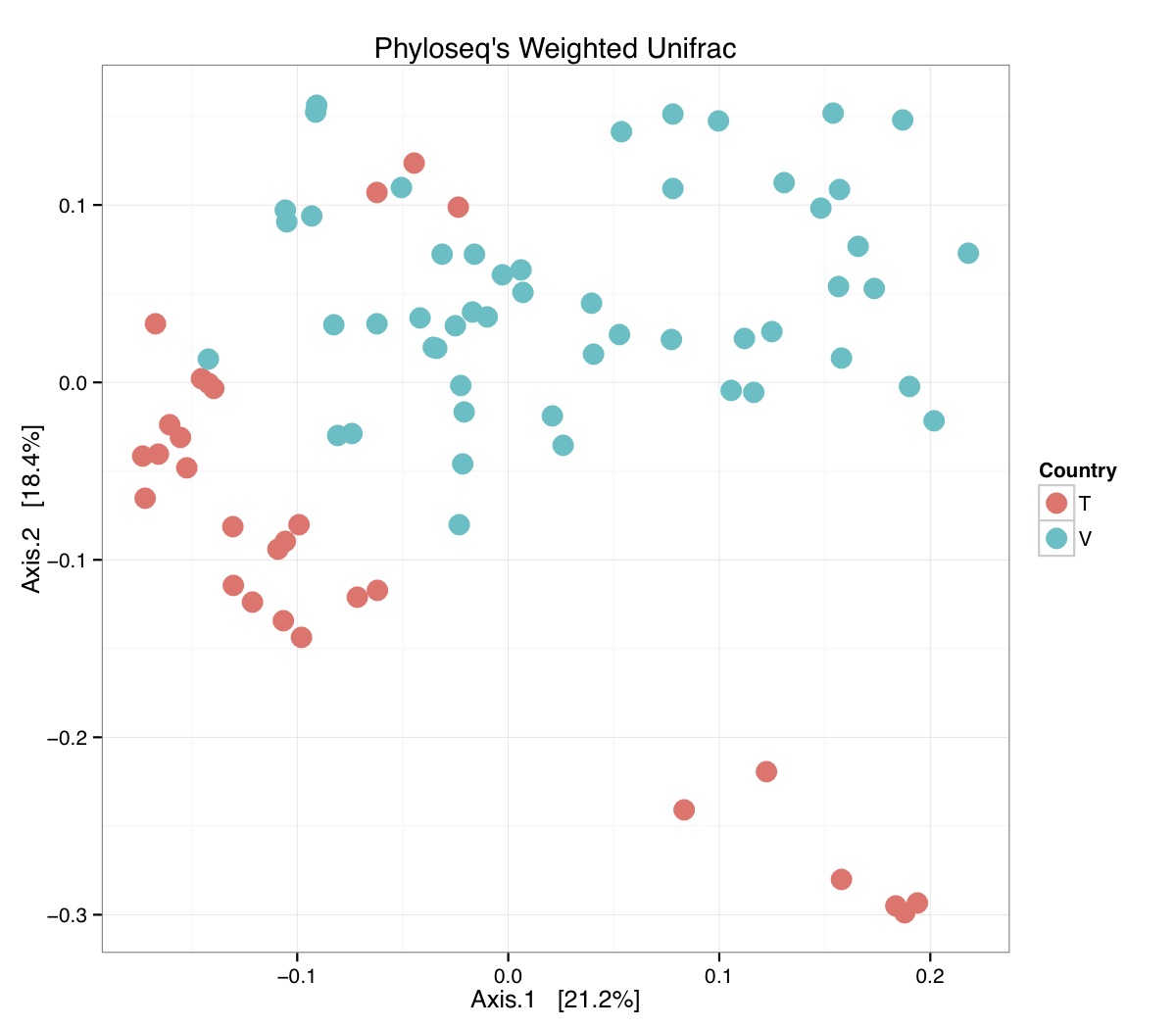
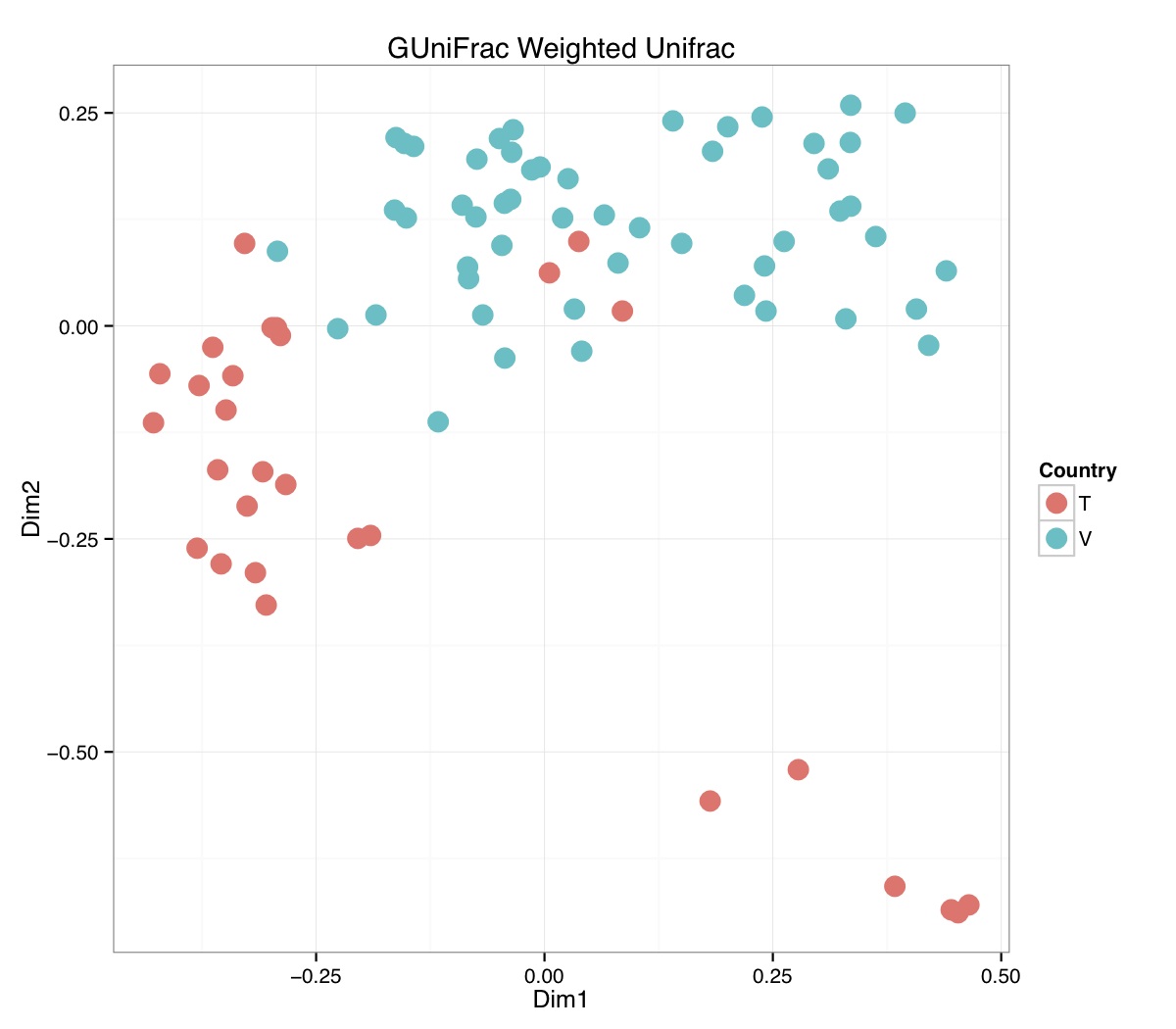
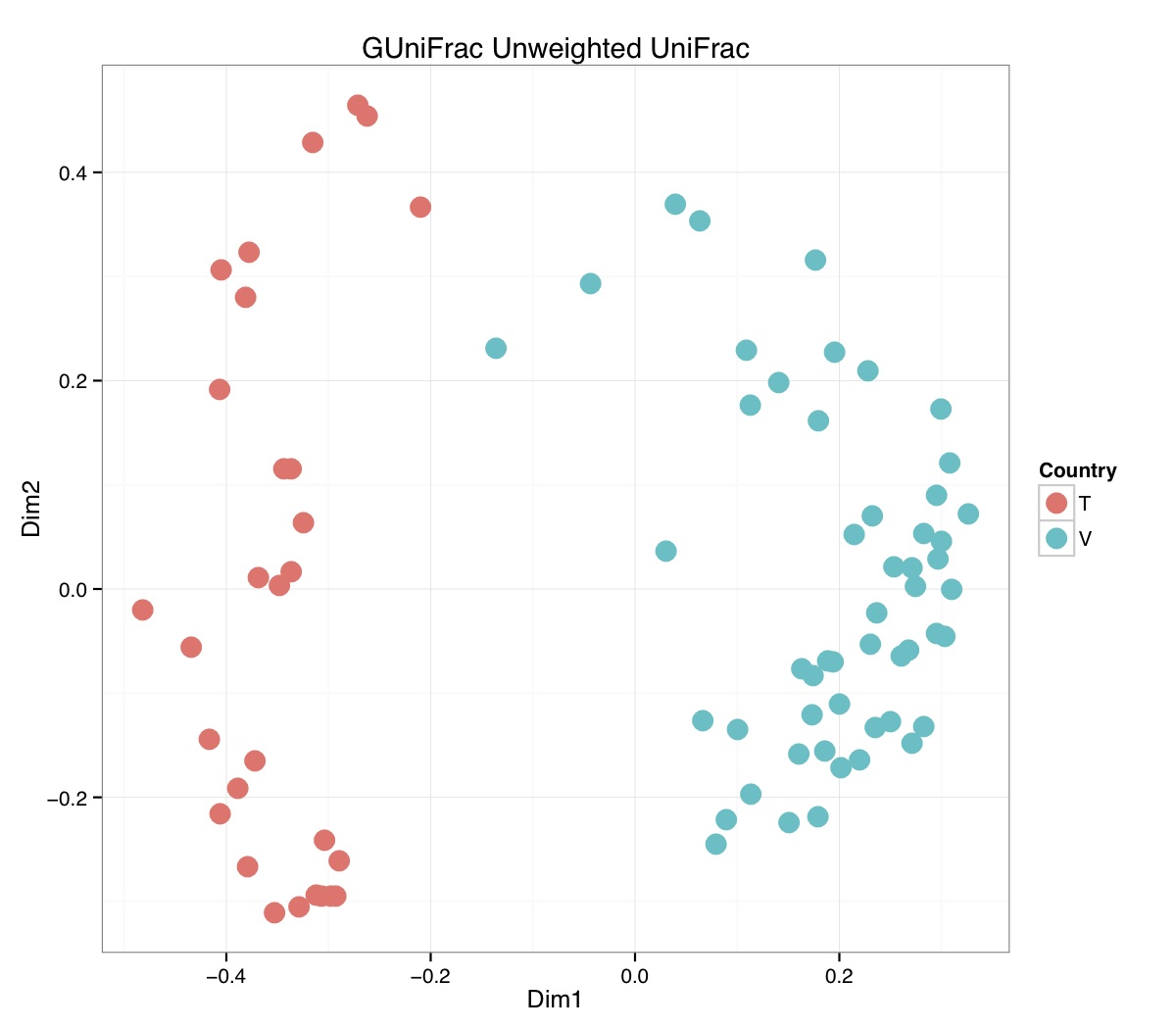
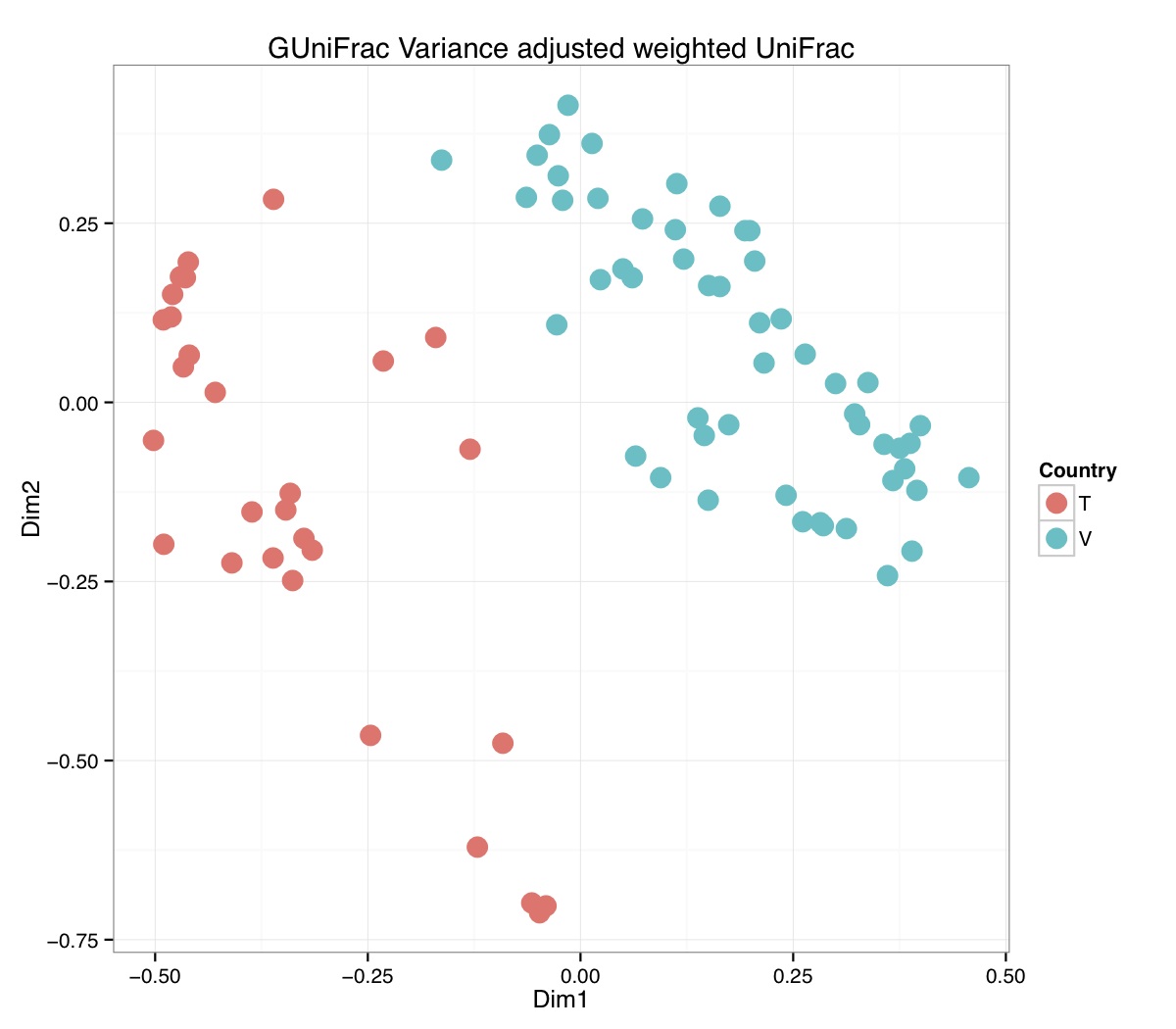
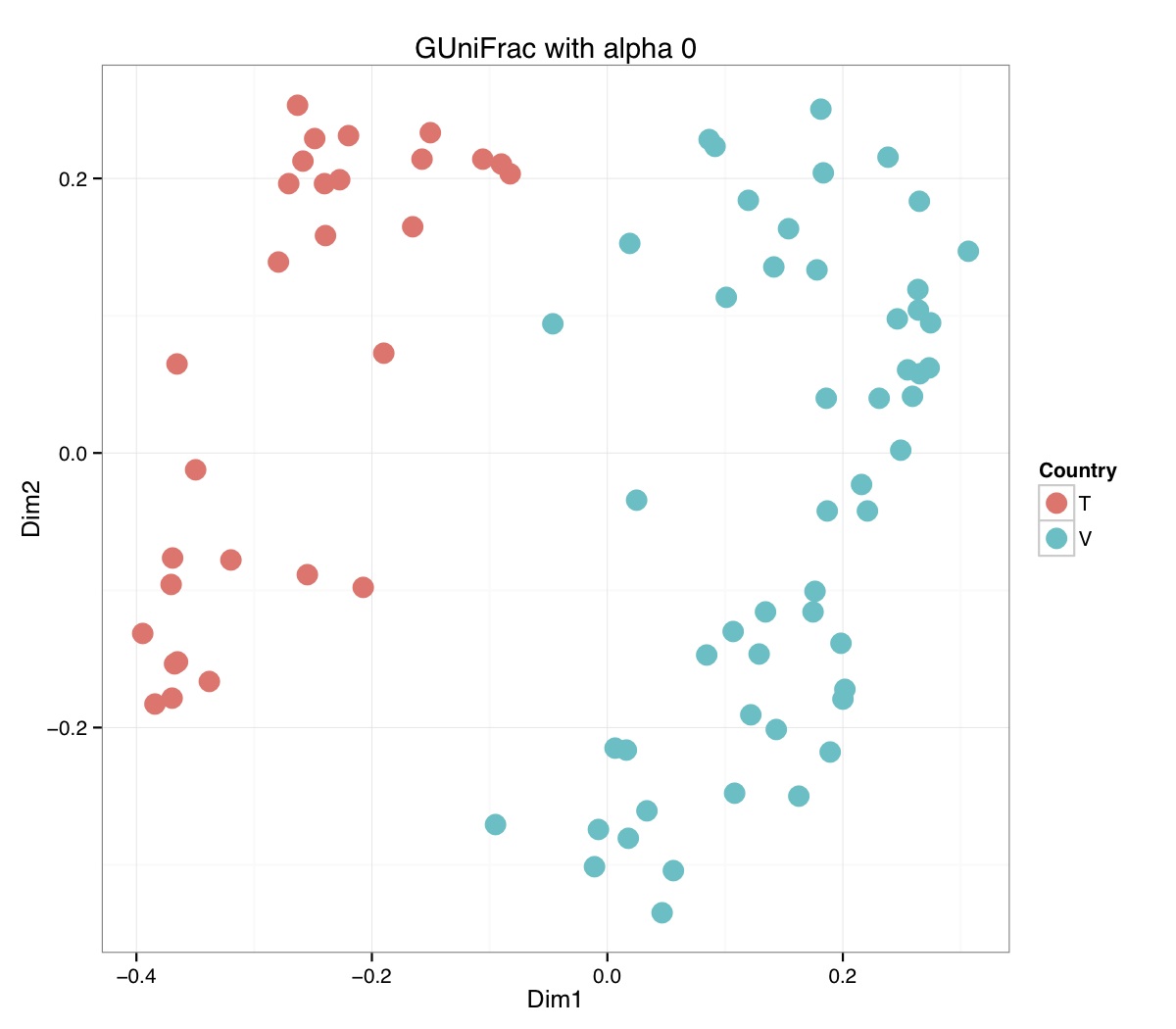
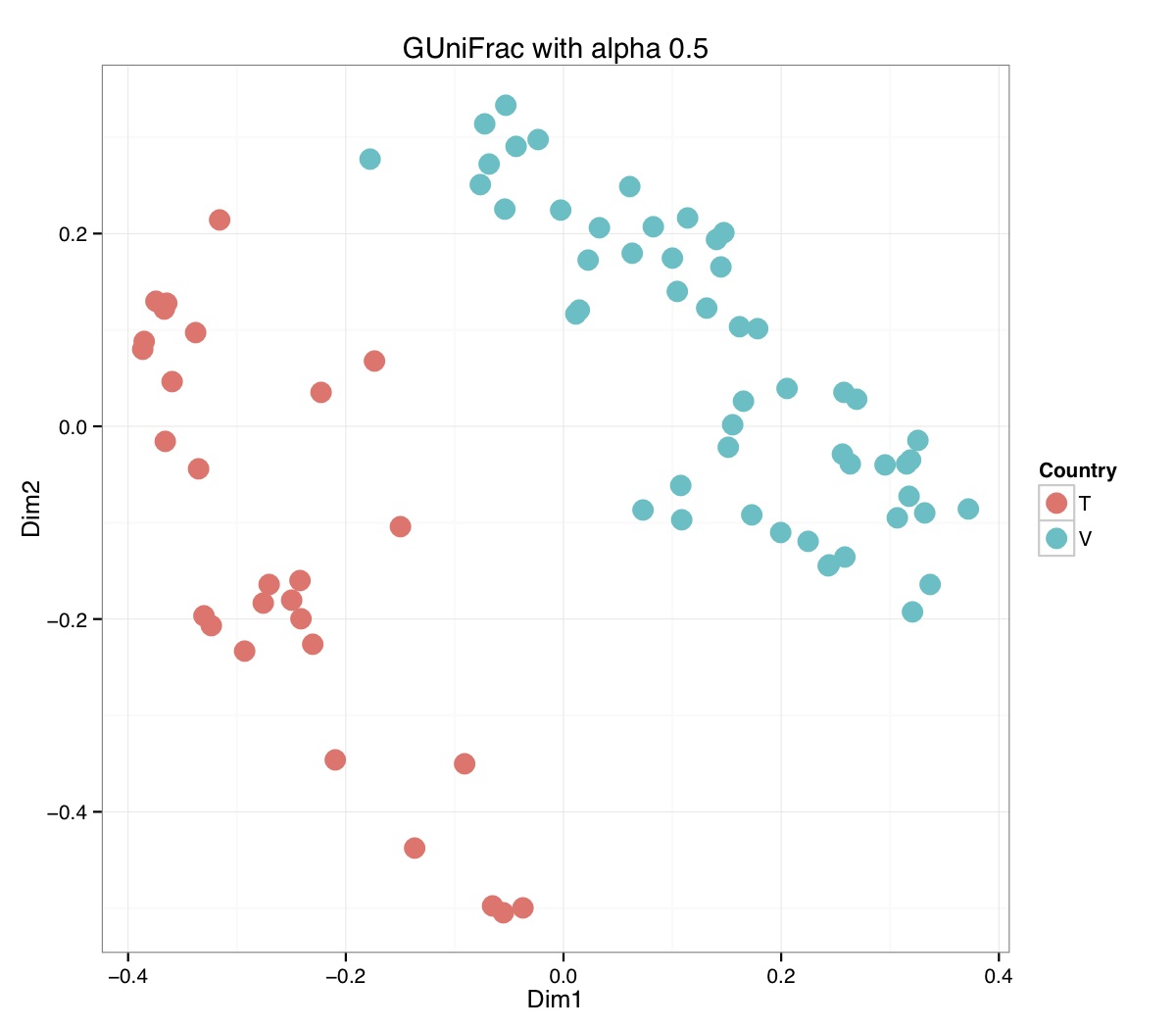
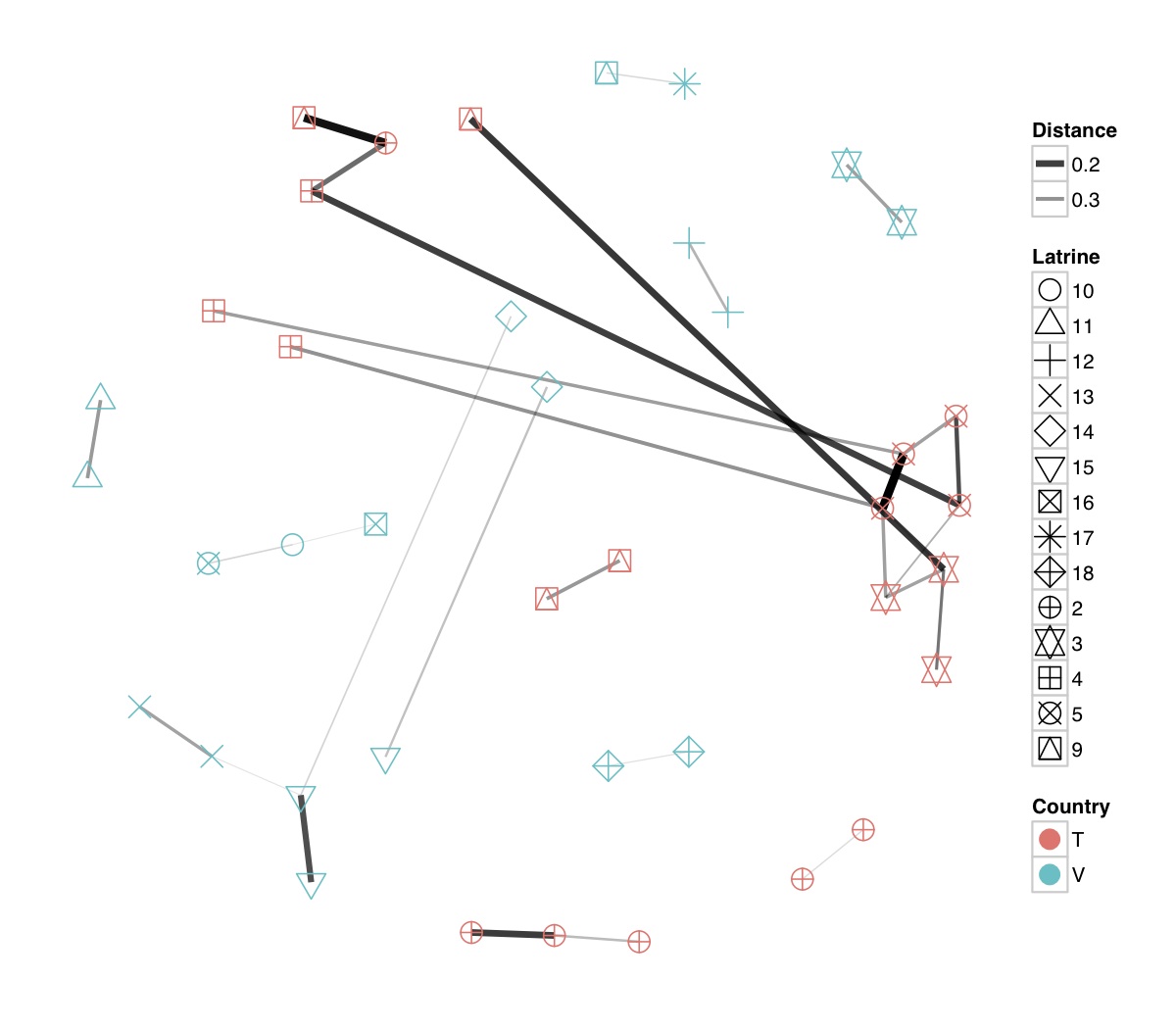
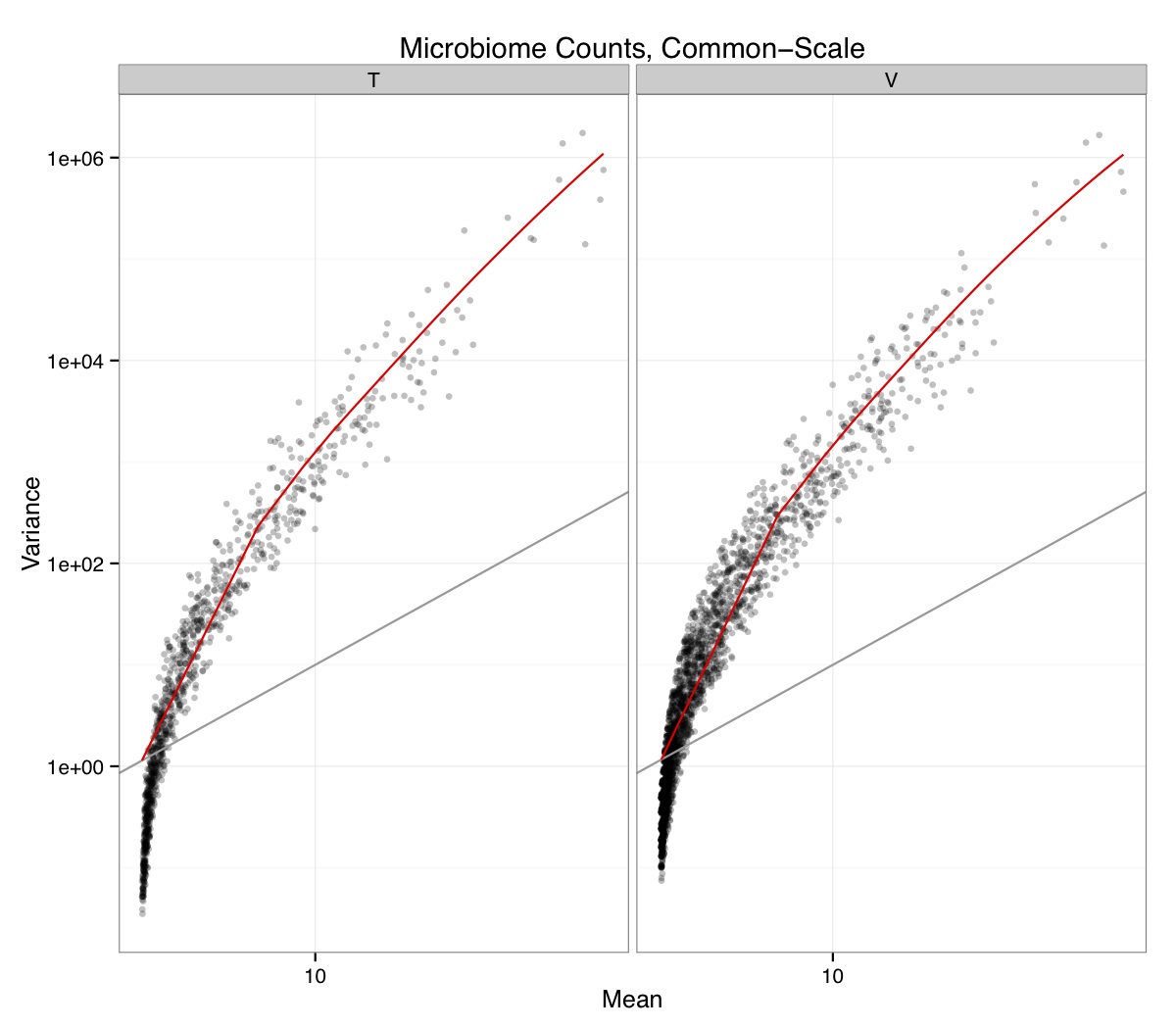
# ============================================================ # Tutorial on Unifrac distances for OTU tables using phyloseq # by Umer Zeeshan Ijaz (http://userweb.eng.gla.ac.uk/umer.ijaz) # ============================================================= library(ggplot2) #Reference: http://joey711.github.io/phyloseq-demo/phyloseq-demo.html #Reference: https://rdp.cme.msu.edu/tutorials/stats/using_rdp_output_with_phyloseq.html #Reference: http://www.daijiang.name/en/2014/05/17/phylogenetic-functional-beta-diversity/ #Reference: http://www.wernerlab.org/teaching/qiime/overview/d #Reference: http://joey711.github.io/waste-not-supplemental/ # Question: # Given an OTU table (All_Good_P2_C03.csv) and corresponding OTU sequences (All_Good_P2_C03.fa), how do you generate phylogenetic tree? # # Solution: # # STEP 1: Aligning sequences # We'll be using a reference alignment to align our sequences. In QIIME, this reference alignment is core_set_aligned.fasta.imputed # and QIIME already knows where it is. The following is done using PyNAST, though alignment can also be done with MUSCLE and Infernal (http://qiime.org/scripts/align_seqs.html). # # $ align_seqs.py -i All_Good_P2_C03.fa -o alignment/ # # STEP 2: Filtering alignment # This alignment contains lots of gaps, and it includes hypervariable regions that make it difficult to build an accurate tree. So, we'll # filter it. Filtering an alignment of 16S rRNA gene sequences can involve a Lane mask. In QIIME, this Lane mask for the GG core is lanemask_in_1s_and_0s # # $ filter_alignment.py -i alignment/All_Good_P2_C03_aligned.fasta -o alignment # # STEP 3: Make tree # make_phylogeny.py script uses the FastTree approximately maximum likelihood program, a good model of evolution for 16S rRNA gene sequences # # $ make_phylogeny.py -i alignment/All_Good_P2_C03_aligned_pfiltered.fasta -o All_Good_P2_C03.tre # # STEP 4: Generate taxonomy using RDP Classifier # # $ java -Xmx1g -jar $RDP_PATH/classifier.jar classify -f filterbyconf -o All_Good_P2_C03_Assignments.txt All_Good_P2_C03.fa # # STEP 5: Finally generate a csv file that can be imported in R # # $ <All_Good_P2_C03_Assignments.txt awk -F"\t" 'BEGIN{print "OTUs,Domain,Phylum,Class,Order,Family,Genus"}{gsub(" ","_",$0);gsub("\"","",$0);print $1","$3","$6","$9","$12","$15","$18}' > All_Good_P2_C03_Taxonomy.csv # abund_table<-read.csv("All_Good_P2_C03.csv",row.names=1,check.names=FALSE) #Transpose the data to have sample names on rows abund_table<-t(abund_table) meta_table<-read.csv("ENV_pitlatrine.csv",row.names=1,check.names=FALSE) #Get grouping information grouping_info<-data.frame(row.names=rownames(meta_table),t(as.data.frame(strsplit(rownames(meta_table),"_")))) colnames(grouping_info)<-c("Country","Latrine","Depth") meta_table<-data.frame(meta_table,grouping_info) #Filter out samples not present in meta_table abund_table<-abund_table[rownames(abund_table) %in% rownames(meta_table),] #Load the tree using ape package library(ape) OTU_tree <- read.tree("All_Good_P2_C03.tre") #Now load the taxonomy OTU_taxonomy<-read.csv("All_Good_P2_C03_Taxonomy.csv",row.names=1,check.names=FALSE) library(phyloseq) #Convert the data to phyloseq format OTU = otu_table(as.matrix(abund_table), taxa_are_rows = FALSE) TAX = tax_table(as.matrix(OTU_taxonomy)) SAM = sample_data(meta_table) physeq<-merge_phyloseq(phyloseq(OTU, TAX),SAM,OTU_tree) #Plot richness p<-plot_richness(physeq, x = "Country", color = "Depth") p<-p+theme_bw() pdf("phyloseq_richness.pdf",width=14) print(p) dev.off() #Now take the subset of the data for latrines at depth 1 and for Prevotellaceae physeq_subset<-subset_taxa(subset_samples(physeq,Depth=="1"),Family=="Prevotellaceae") p <- plot_tree(physeq_subset, color = "Country", label.tips = "Genus", size = "abundance",text.size=2) pdf("phyloseq_tree.pdf",width=8) print(p) dev.off() #We now take the 500 most abundant Taxa physeq_subset<-prune_taxa(names(sort(taxa_sums(physeq), TRUE)[1:500]), physeq) #Plot heatmap p<-plot_heatmap(physeq_subset, method=NULL, sample.label="Country", taxa.label="Class") pdf("phyloseq_heatmap.pdf",width=8) print(p) dev.off() #Plot bar p<-plot_bar(physeq_subset, x="Country", fill="Phylum")+theme_bw() pdf("phyloseq_bar.pdf",width=8) print(p) dev.off() #Make separate panel for each country p<-plot_bar(physeq_subset, x="Phylum", fill="Phylum",facet_grid=~Country)+theme_bw() p<-p+theme(axis.text.x=element_text(angle=90,hjust=1,vjust=0.5)) pdf("phyloseq_bar_separate.pdf",width=8) print(p) dev.off() #Make an PCoA ordination plot based on abundance based unifrac distances with the following commands ord <- ordinate(physeq_subset, method="PCoA", distance="unifrac", weighted=TRUE) p <- plot_ordination(physeq_subset, ord, color="Country",title="Phyloseq's Weighted Unifrac") p <- p + geom_point(size=5) + theme_bw() pdf("phyloseq_unifrac.pdf",width=8) print(p) dev.off() #The GUniFrac package can also be used to calculate unifrac distances and has additional features. #Unifrac distances are traditionally calculated on either presence/absence data, or abundance data. #The former can be affected by PCR and sequencing errors leading to a high number of spurious and #usually rare OTUs, and the latter can give undue weight to the more abundant OTUs. #GUniFrac's methods include use of a parameter alpha that controls the weight given to abundant OTUs #and also a means of adjusting variances. library(GUniFrac) library(phangorn) #The function GUniFrac requires a rooted tree, but unlike phyloseq's ordination function #will not try to root an unrooted one. We will apply mid-point rooting with the midpoint function #from the phangorn package unifracs <- GUniFrac(as.matrix(otu_table(physeq_subset)), midpoint(phy_tree(physeq_subset)), alpha = c(0, 0.5, 1))$unifracs # We can extract a variety of distance matrices with different weightings. dw <- unifracs[, , "d_1"] # Weighted UniFrac du <- unifracs[, , "d_UW"] # Unweighted UniFrac dv <- unifracs[, , "d_VAW"] # Variance adjusted weighted UniFrac d0 <- unifracs[, , "d_0"] # GUniFrac with alpha 0 d5 <- unifracs[, , "d_0.5"] # GUniFrac with alpha 0.5 # use vegan's cmdscale function to make a PCoA ordination from a distance matrix. pcoa <- cmdscale(dw, k = nrow(as.matrix(otu_table(physeq_subset))) - 1, eig = TRUE, add = TRUE) p<-plot_ordination(physeq_subset, pcoa, color="Country", title="GUniFrac Weighted Unifrac") + geom_point(size=5)+ theme_bw() pdf("GUniFrac_weighted.pdf",width=8) print(p) dev.off() pcoa <- cmdscale(du, k = nrow(as.matrix(otu_table(physeq_subset))) - 1, eig = TRUE, add = TRUE) p<-plot_ordination(physeq_subset, pcoa, color="Country", title="GUniFrac Unweighted UniFrac") + geom_point(size=5)+ theme_bw() pdf("GUniFrac_unweighted.pdf",width=8) print(p) dev.off() pcoa <- cmdscale(dv, k = nrow(as.matrix(otu_table(physeq_subset))) - 1, eig = TRUE, add = TRUE) p<-plot_ordination(physeq_subset, pcoa, color="Country", title="GUniFrac Variance adjusted weighted UniFrac") + geom_point(size=5)+ theme_bw() pdf("GUniFrac_variance.pdf",width=8) print(p) dev.off() pcoa <- cmdscale(d0, k = nrow(as.matrix(otu_table(physeq_subset))) - 1, eig = TRUE, add = TRUE) p<-plot_ordination(physeq_subset, pcoa, color="Country", title="GUniFrac with alpha 0") + geom_point(size=5)+ theme_bw() pdf("GUniFrac_alpha0.pdf",width=8) print(p) dev.off() pcoa <- cmdscale(d5, k = nrow(as.matrix(otu_table(physeq_subset))) - 1, eig = TRUE, add = TRUE) p<-plot_ordination(physeq_subset, pcoa, color="Country", title="GUniFrac with alpha 0.5") + geom_point(size=5)+ theme_bw() pdf("GUniFrac_alpha0.5.pdf",width=8) print(p) dev.off() #Permanova - Distance based multivariate analysis of variance adonis(as.dist(d5) ~ as.matrix(sample_data(physeq_subset)[,"Country"])) # > adonis(as.dist(d5) ~ as.matrix(sample_data(physeq_subset)[,"Country"])) # # Call: # adonis(formula = as.dist(d5) ~ as.matrix(sample_data(physeq_subset)[, "Country"])) # # Permutation: free # Number of permutations: 999 # # Terms added sequentially (first to last) # # Df SumsOfSqs MeanSqs F.Model R2 Pr(>F) # as.matrix(sample_data(physeq_subset)[, "Country"]) 1 3.7531 3.7531 14.558 0.1556 0.001 *** # Residuals 79 20.3666 0.2578 0.8444 # Total 80 24.1198 1.0000 # --- # Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 #Each distance measure is most powerful in detecting only a certain scenario. When multiple distance #matrices are available, separate tests using each distance matrix will lead to loss of power due to #multiple testing correction. Combing the distance matrices in a single test will improve power. #PermanovaG combines multiple distance matrices by taking the maximum of pseudo-F statistics #for each distance matrix. Significance is assessed by permutation. PermanovaG(unifracs[, , c("d_0", "d_0.5", "d_1")] ~ as.matrix(sample_data(physeq_subset)[,"Country"])) # > PermanovaG(unifracs[, , c("d_0", "d_0.5", "d_1")] ~ as.matrix(sample_data(physeq_subset)[,"Country"])) # $aov.tab # F.Model p.value # as.matrix(sample_data(physeq_subset)[, "Country"]) 14.55806 0.001 #Make a network based on GUniFrac with alpha 0.5 p<-plot_net(physeq_subset, distance=as.dist(d5), maxdist = 0.4, color = "Country", shape="Latrine") p<-p + scale_shape_manual(values=seq(1,15)) pdf("phyloseq_network.pdf",width=8) print(p) dev.off() #Reference:http://joey711.github.io/waste-not-supplemental/dispersion-survey/dispersion.html #Justification that microbiome count data from amplicon sequencing experiments can be treated #with the same procedures as for differential expression data from RNA-Seq experiments, #that microbiome data is overdispersed, that is, has an effectively non-zero value for #dispersion parameter in the Negative Binomial generalization of Poisson discrete counts. #UZI has modified the deseq_var from above as there was a bug deseq_var = function(physeq, bioreps) { require("DESeq") require("plyr") # physeq = GP bioreps = 'BioReps' biorep_levels = levels(factor(get_variable(physeq, bioreps))) ldfs = lapply(biorep_levels, function(L, physeq, bioreps) { # L = biorep_levels[1] whsa = as(get_variable(physeq, bioreps), "character") %in% L # Skip if there are 2 or fewer instances among BioReps Prob don't want to # assume very much from estimates of variance for which N<=2 if (sum(whsa) <= 2) { return(NULL) } ps1 = prune_samples(whsa, physeq) # Prune the 'zero' OTUs from this sample set. ps1 = prune_taxa(taxa_sums(ps1) > 5, physeq) # Coerce to matrix, round up to nearest integer just in case x = ceiling(as(otu_table(ps1), "matrix")) # Add 1 to everything to avoid log-0 problems x = x + 1 #UZI put t(x) instead of x in the following cds = newCountDataSet(countData = t(x), conditions = get_variable(physeq, bioreps)) # Estimate Size factors cds = estimateSizeFactors(cds, locfunc = median) # Estimate Dispersions, DESeq cds = estimateDispersions(cds, method = "pooled", sharingMode = "fit-only", fitType = "local") # get the scale-normalized mean and variance df = getBaseMeansAndVariances(counts(cds), sizeFactors(cds)) colnames(df) <- c("Mean", "Variance") ## Calculate Mean and Variance for each OTU after rarefying Use a non-random ## definition of 'rarefying' ps1r = transform_sample_counts(ps1, function(x, Smin = min(sample_sums(ps1), na.rm = TRUE)) { round(x * Smin/sum(x), digits = 0) }) ps1r = prune_taxa(taxa_sums(ps1r) > 5, ps1r) rdf = data.frame(Mean = apply(otu_table(ps1r), 1, mean), Variance = apply(otu_table(ps1r), 1, var)) # Return results, including cds and 'rarefied' mean-var return(list(df = df, cds = cds, rdf = rdf)) }, physeq, bioreps) names(ldfs) <- biorep_levels # Remove the elements that were skipped. ldfs <- ldfs[!sapply(ldfs, is.null)] # Group into data.frames of variance estimates df = ldply(ldfs, function(L) { data.frame(L$df, OTUID = rownames(L$df)) }) rdf = ldply(ldfs, function(L) { data.frame(L$rdf, OTUID = rownames(L$rdf)) }) colnames(df)[1] <- bioreps colnames(rdf)[1] <- bioreps # Remove values that are zero for both df <- df[!(df$Mean == 0 & df$Variance == 0), ] rdf <- rdf[!(rdf$Mean == 0 & rdf$Variance == 0), ] # Create a `data.frame` of the DESeq predicted. deseqfitdf = ldply(ldfs, function(x) { df = x$df xg = seq(range(df$Mean)[1], range(df$Mean)[2], length.out = 100) fitVar = xg + fitInfo(x$cds)$dispFun(xg) * xg^2 return(data.frame(Mean = xg, Variance = fitVar)) }) colnames(deseqfitdf)[1] <- bioreps return(list(datadf = df, fitdf = deseqfitdf, raredf = rdf)) } restrovar = deseq_var(physeq, "Country") #UZI has taken facet_wrap(~BioReps) out from the following plot_dispersion <- function(datadf, fitdf, title = "Microbiome Counts, Common-Scale", xbreaks = c(10, 1000, 1e+05)) { require("ggplot2") p = ggplot(datadf, aes(Mean, Variance)) + geom_point(alpha = 0.25, size = 1.5) p = p + scale_y_log10() p = p + scale_x_log10(breaks = xbreaks, labels = xbreaks) p = p + geom_abline(intercept = 0, slope = 1, linetype = 1, size = 0.5, color = "gray60") p = p + geom_line(data = fitdf, color = "red", linetype = 1, size = 0.5) p = p + ggtitle(title) return(p) } p<-plot_dispersion(restrovar$datadf, restrovar$fitdf)+facet_wrap(~Country)+theme_bw() pdf("phyloseq_dispersion.pdf",width=8) print(p) dev.off()