Shell Utilities for QC
by Umer Zeeshan Ijaz and Chris Quince
We have written shell utilities that emulate the functionality of FastQC, a popular software that allows quality control checks on raw sequence data coming from high throughput sequencing pipelines. The sheer strength of these shell utilities comes from the powerful perl one-liners that have resulted in very compact scripts. For visualisation, we use gplot, a wrapper around gnuplot, that facilitates us to pipe quantitative data directly into gnuplot without the need of generating temporary files.
Dependencies
emboss, Download from here
gplot, Install using git clone https://github.com/RhysU/gplot.git
Statistics::Descriptive, Install using sudo perl -MCPAN -e shell install Statistics::Descriptive
Available Utilities
perbase_quality_FASTQ.sh: Per-base quality score for FASTQ file
average_quality_hist_FASTQ.sh: Average quality distribution for FASTQ file
duplication_hist_FAST[Q/A].sh: Duplication distribution for FASTQ/FASTA file
length_distribution_FAST[Q/A].sh: Length distribution for FASTQ/FASTA file
average_GC_hist_FAST[Q/A].sh: Average GC distribution for FASTQ/FASTA file
perbase_seqcontent_FAST[Q/A].sh: Per-base sequence content for FASTQ/FASTA file
perbase_GCcontent_FAST[Q/A].sh: Per-base GC content for FASTQ/FASTA file
perbase_ATcontent_FAST[Q/A].sh: Per-base AT content for FASTQ/FASTA file
perbase_Ncontent_FAST[Q/A].sh: Per-base N content for FASTQ/FASTA file
top_kmer_FAST[Q/A].sh: Top N Kmers for FASTQ/FASTA file
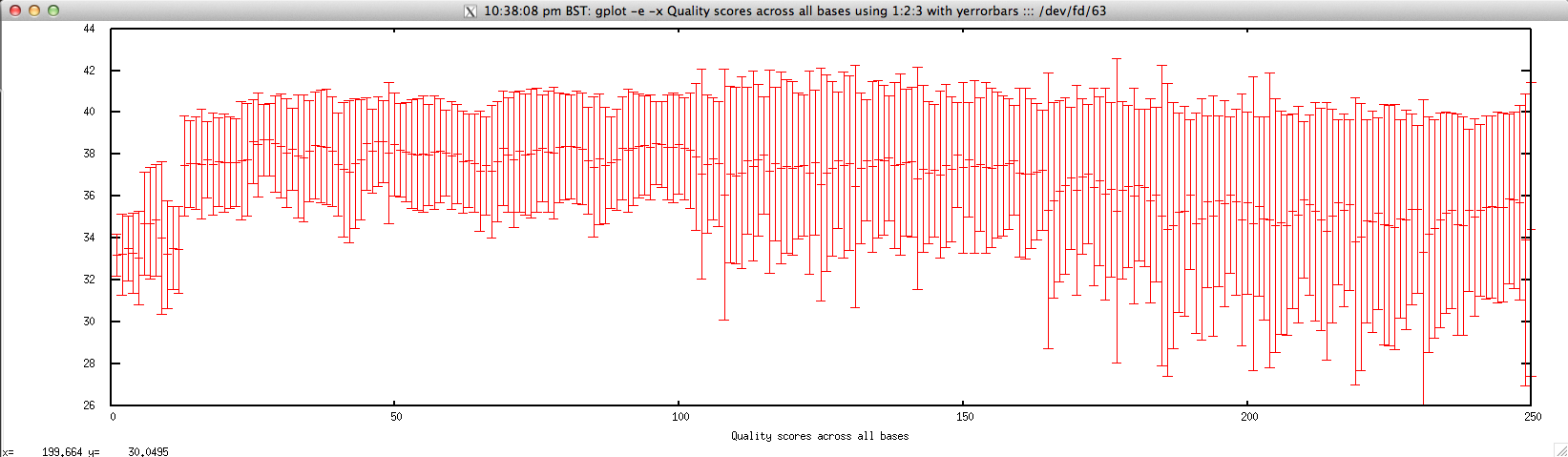
Usage: ./perbase_quality_FASTQ.sh file.fastq
#!/bin/bash
gplot -e -x "Quality scores across all bases" using 1:2:3 with yerrorbars ::: <(cat $1 | \
perl -MStatistics::Descriptive -lne 'push @a, $_; @a = @a[@a-4..$#a]; if ($. % 4 == 0){
chomp($a[3]);
$max_j=0;
$j=0;
++$i;
map{$r{$i.":".++$j}=(ord($_)-33)}split("",$a[3]);
if($j>$max_j){
$max_j=$j;}}
}{
for($jj=1;$jj<=$max_j;$jj++){
@m=();
for($ii=1;$ii<=$i;$ii++){
unless(not defined $r{$ii.":".$jj})
{push @m,$r{$ii.":".$jj}}}
$s=Statistics::Descriptive::Full->new();
$s->add_data(@m);
print($jj."\t".$s->mean()."\t".$s->standard_deviation())}')
Output: ./perbase_quality_FASTQ.sh forward.fastq
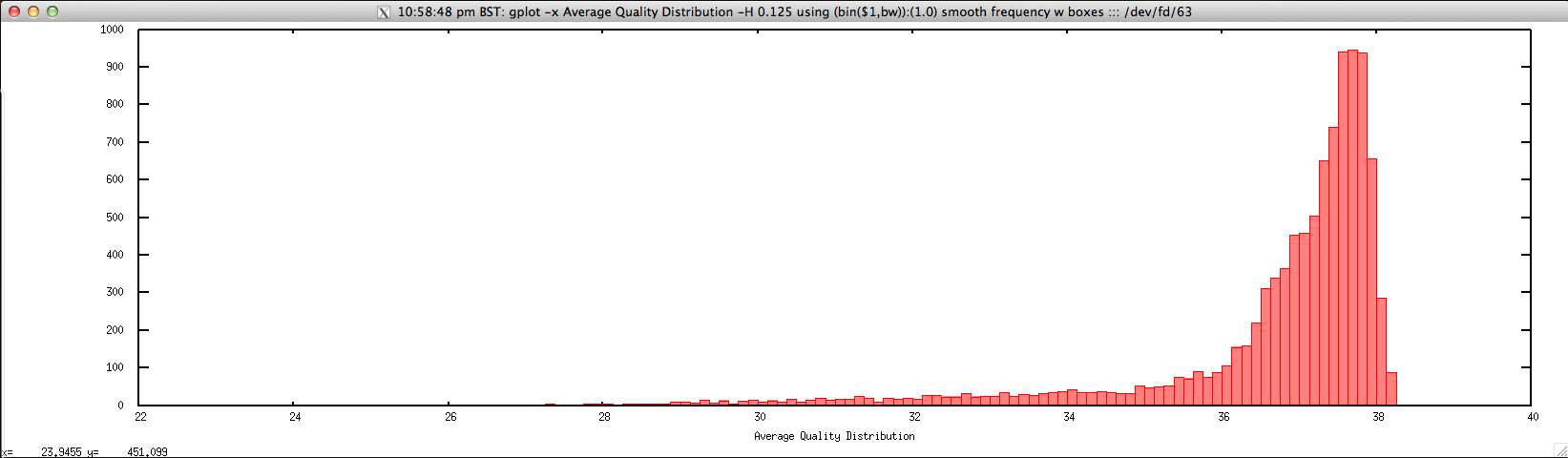
 Usage: ./average_quality_hist_FASTQ.sh file.fastq histogram_bin_size
Usage: ./average_quality_hist_FASTQ.sh file.fastq histogram_bin_size
#!/bin/bash
gplot -x "Average Quality Distribution" -H $2 using '(bin($1,bw)):(1.0)' smooth frequency w boxes ::: <(cat $1 | \
perl -lne 'push @a, $_; @a = @a[@a-4..$#a]; if ($. % 4 == 0){
chomp($a[3]);
$s=0;
map{$s+=ord($_)-33}split("",$a[3]);
print $s/$a[3]=~y===c}')
Output: ./average_quality_hist_FASTQ.sh forward.fastq 0.125
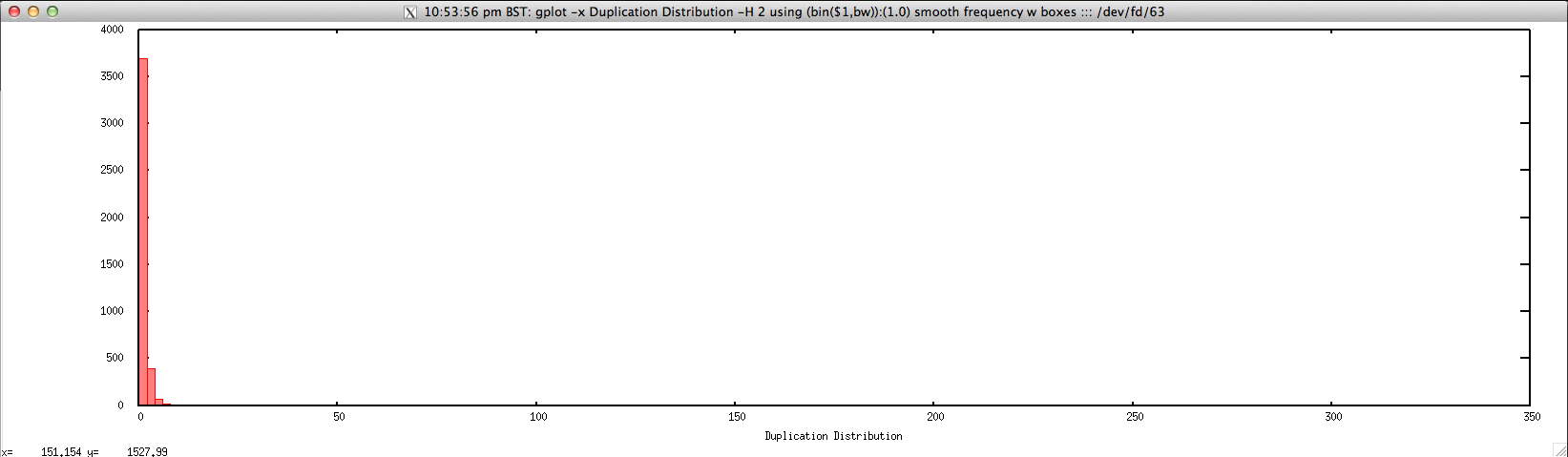
 Usage: ./duplication_hist_FASTQ.sh file.fastq histogram_bin_size
Usage: ./duplication_hist_FASTQ.sh file.fastq histogram_bin_size
#!/bin/bash
gplot -x "Duplication Distribution" -H $2 using '(bin($1,bw)):(1.0)' smooth frequency w boxes ::: <(cat $1 | \
perl -ne 'push @a, $_; @a = @a[@a-4..$#a]; if ($. % 4 == 0){
chomp($a[1]);
$d{uc($a[1])}++;}
END {
foreach my $n (sort{ $d{$b} <=> $d{$a} } keys %d){
print $d{$n}."\n"}}')
Usage ./duplication_hist_FASTA.sh file.fasta histogram_bin_size
#!/bin/bash
gplot -x "Duplication Distribution" -H $2 using '(bin($1,bw)):(1.0)' smooth frequency w boxes ::: <(cat $1 | \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -ne 'push @a, $_; @a = @a[@a-2..$#a]; if ($. % 2 == 0){
chomp($a[1]);
$d{uc($a[1])}++;}
END {
foreach my $n (sort{ $d{$b} <=> $d{$a} } keys %d){
print $d{$n}."\n"}}')
Output: ./duplication_hist_FASTQ.sh forward.fastq 2
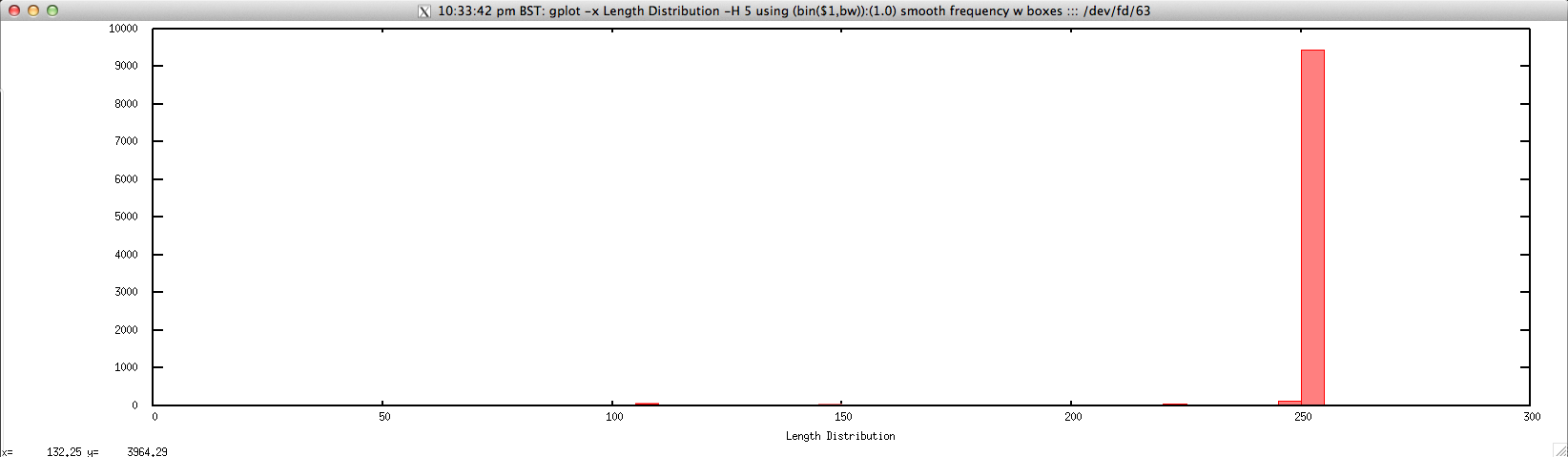
 Usage: ./length_distribution_FASTQ.sh file.fastq histogram_bin_size
Usage: ./length_distribution_FASTQ.sh file.fastq histogram_bin_size
#!/bin/bash
gplot -x "Length Distribution" -H $2 using '(bin($1,bw)):(1.0)' smooth frequency w boxes ::: <(cat $1 | \
perl -lne 'push @a, $_; @a = @a[@a-4..$#a]; if ($. % 4 == 0){
chomp($a[1]);
print ($a[1]=~y===c)}')
Usage ./length_distribution_FASTA.sh file.fasta histogram_bin_size
#!/bin/bash
gplot -x "Length Distribution" -H $2 using '(bin($1,bw)):(1.0)' smooth frequency w boxes ::: <(cat $1 | \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -lne 'print $_=y///c if !/^>/')
Output: ./length_distribution_FASTQ.sh forward.fastq 5
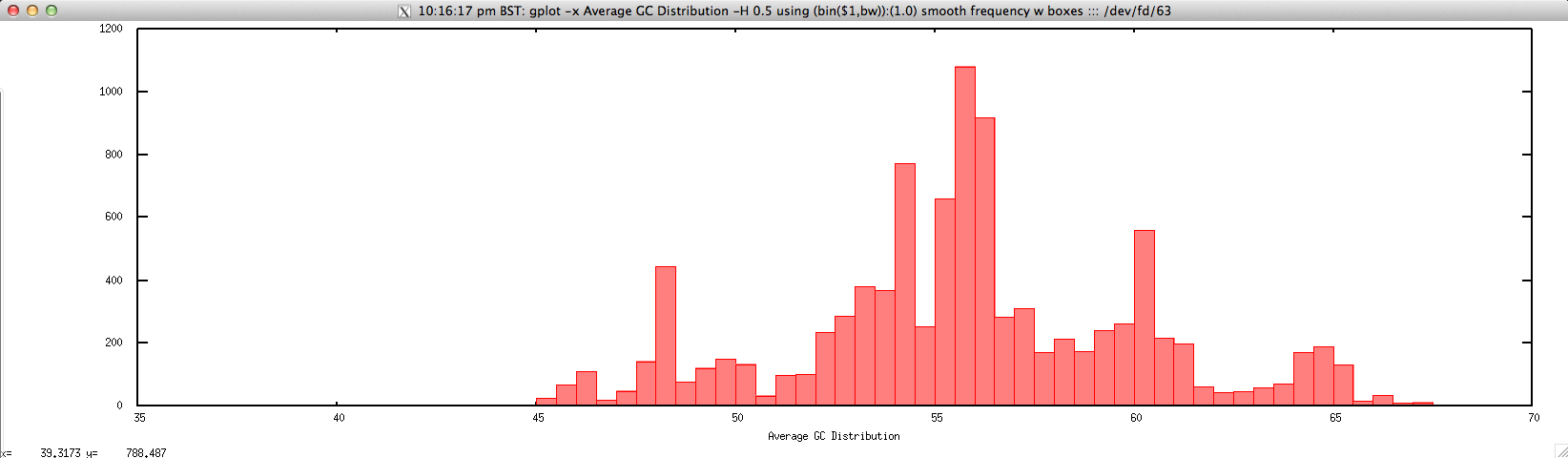
 Usage: ./average_GC_hist_FASTQ.sh histogram_bin_size
Usage: ./average_GC_hist_FASTQ.sh histogram_bin_size
#!/bin/bash
gplot -x "Average GC Distribution" -H $2 using '(bin($1,bw)):(1.0)' smooth frequency w boxes ::: <(cat $1 | \
perl -lne 'push @a, $_; @a = @a[@a-4..$#a]; if ($. % 4 == 0){
chomp($a[1]);
print (($a[1]=~tr/GC//)/($a[1]=~y===c)*100)}')
Usage: ./average_GC_hist_FASTA.sh histogram_bin_size
#!/bin/bash
gplot -x "Average GC Distribution" -H $2 using '(bin($1,bw)):(1.0)' smooth frequency w boxes ::: <(cat $1 | \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -lne 'push @a, $_; @a = @a[@a-2..$#a]; if ($. % 2 == 0){
chomp($a[1]);
print (($a[1]=~tr/GC//)/($a[1]=~y===c)*100)}')
Output: ./average_GC_hist_FASTQ.sh forward.fastq 0.5
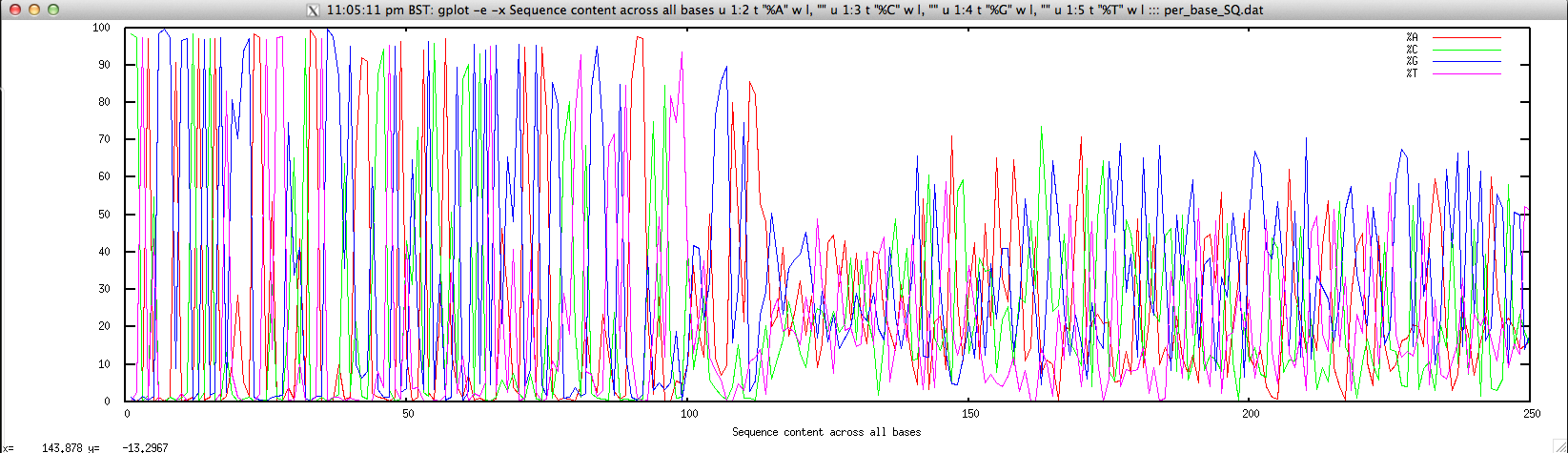
 Usage: ./perbase_seqcontent_FASTQ.sh file.fastq
Usage: ./perbase_seqcontent_FASTQ.sh file.fastq
#!/bin/bash
cat $1 | perl -lne 'push @a, $_; @a = @a[@a-4..$#a]; if ($. % 4 == 0){
chomp($a[1]);
$max_j=0;
$j=0;++$i;
map{$r{$i.":".++$j}=$_}split("",$a[1]);
if($j>$max_j){
$max_j=$j;}}
}{
for($jj=1;$jj<=$max_j;$jj++){
@m=();
for($ii=1;$ii<=$i;$ii++){
unless(not defined $r{$ii.":".$jj}){
push @m,$r{$ii.":".$jj}}}
$s=join("",@m);
$l=($s=~y===c);
print ($jj."\t".((($s=~tr/A//)/$l)*100)."\t".((($s=~tr/C//)/$l)*100)."\t".((($s=~tr/G//)/$l)*100)."\t".((($s=~tr/T//)/$l)*100))}' > per_base_SQ.dat; \
gplot -e -x "Sequence content across all bases" u 1:2 t \"%A\" w l, \"\" u 1:3 t \"%C\" w l, \"\" u 1:4 t \"%G\" w l, \"\" u 1:5 t \"%T\" w l ::: per_base_SQ.dat; \
rm per_base_SQ.dat
Usage: ./perbase_seqcontent_FASTA.sh file.fasta
#!/bin/bash
cat $1 | perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -lne 'push @a, $_; @a = @a[@a-2..$#a]; if ($. % 2 == 0){
chomp($a[1]);
$max_j=0;
$j=0;++$i;
map{$r{$i.":".++$j}=$_}split("",$a[1]);
if($j>$max_j){
$max_j=$j;}}
}{
for($jj=1;$jj<=$max_j;$jj++){
@m=();
for($ii=1;$ii<=$i;$ii++){
unless(not defined $r{$ii.":".$jj}){
push @m,$r{$ii.":".$jj}}}
$s=join("",@m);
$l=($s=~y===c);
print ($jj."\t".((($s=~tr/A//)/$l)*100)."\t".((($s=~tr/C//)/$l)*100)."\t".((($s=~tr/G//)/$l)*100)."\t".((($s=~tr/T//)/$l)*100))}' > per_base_SQ.dat; \
gplot -e -x "Sequence content across all bases" u 1:2 t \"%A\" w l, \"\" u 1:3 t \"%C\" w l, \"\" u 1:4 t \"%G\" w l, \"\" u 1:5 t \"%T\" w l ::: per_base_SQ.dat; \
rm per_base_SQ.dat
Output: ./perbase_seqcontent_FASTQ.sh forward.fastq
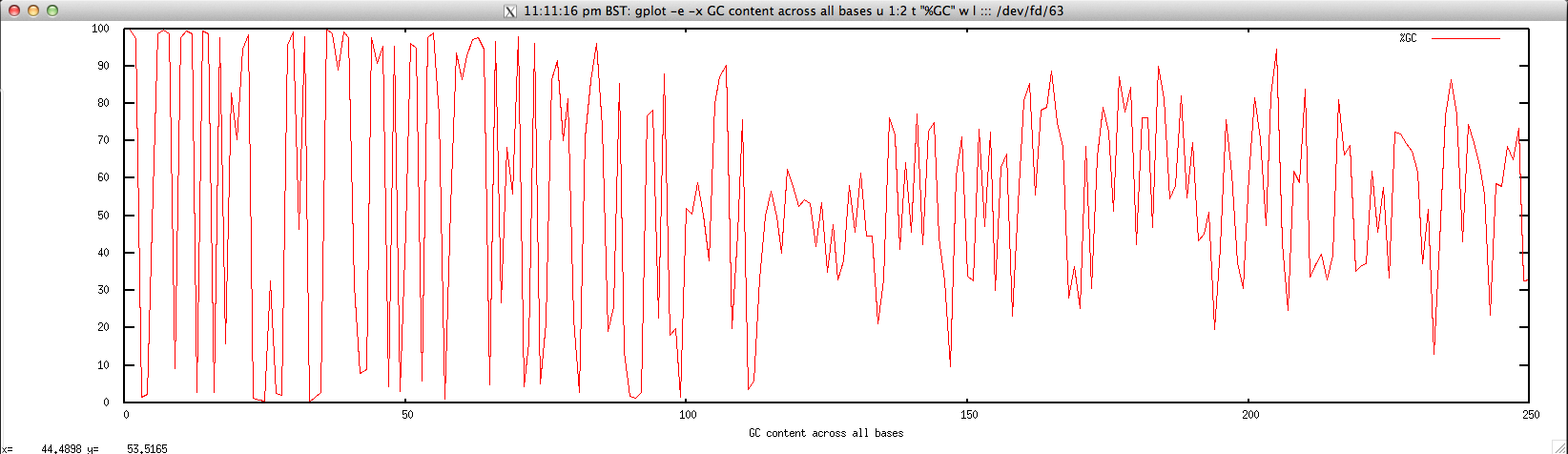
 Usage: ./perbase_GCcontent_FASTQ.sh file.fastq
Usage: ./perbase_GCcontent_FASTQ.sh file.fastq
#!/bin/bash
gplot -e -x "GC content across all bases" u 1:2 t \"%GC\" w l ::: <(cat $1 | \
perl -lne 'push @a, $_; @a = @a[@a-4..$#a]; if ($. % 4 == 0){
chomp($a[1]);
$max_j=0;
$j=0;++$i;
map{$r{$i.":".++$j}=$_}split("",$a[1]);
if($j>$max_j){
$max_j=$j;}}
}{
for($jj=1;$jj<=$max_j;$jj++){
@m=();
for($ii=1;$ii<=$i;$ii++){
unless(not defined $r{$ii.":".$jj}){
push @m,$r{$ii.":".$jj}}}
$s=join("",@m);
$l=($s=~y===c);
print ($jj."\t".((($s=~tr/GC//)/$l)*100))}')
Usage: ./perbase_GCcontent_FASTA.sh file.fasta
#!/bin/bash
gplot -e -x "GC content across all bases" u 1:2 t \"%GC\" w l ::: <(cat $1 | \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -lne 'push @a, $_; @a = @a[@a-2..$#a]; if ($. % 2 == 0){
chomp($a[1]);
$max_j=0;
$j=0;++$i;
map{$r{$i.":".++$j}=$_}split("",$a[1]);
if($j>$max_j){
$max_j=$j;}}
}{
for($jj=1;$jj<=$max_j;$jj++){
@m=();
for($ii=1;$ii<=$i;$ii++){
unless(not defined $r{$ii.":".$jj}){
push @m,$r{$ii.":".$jj}}}
$s=join("",@m);
$l=($s=~y===c);
print ($jj."\t".((($s=~tr/GC//)/$l)*100))}')
Output: ./perbase_GCcontent_FASTQ.sh forward.fastq
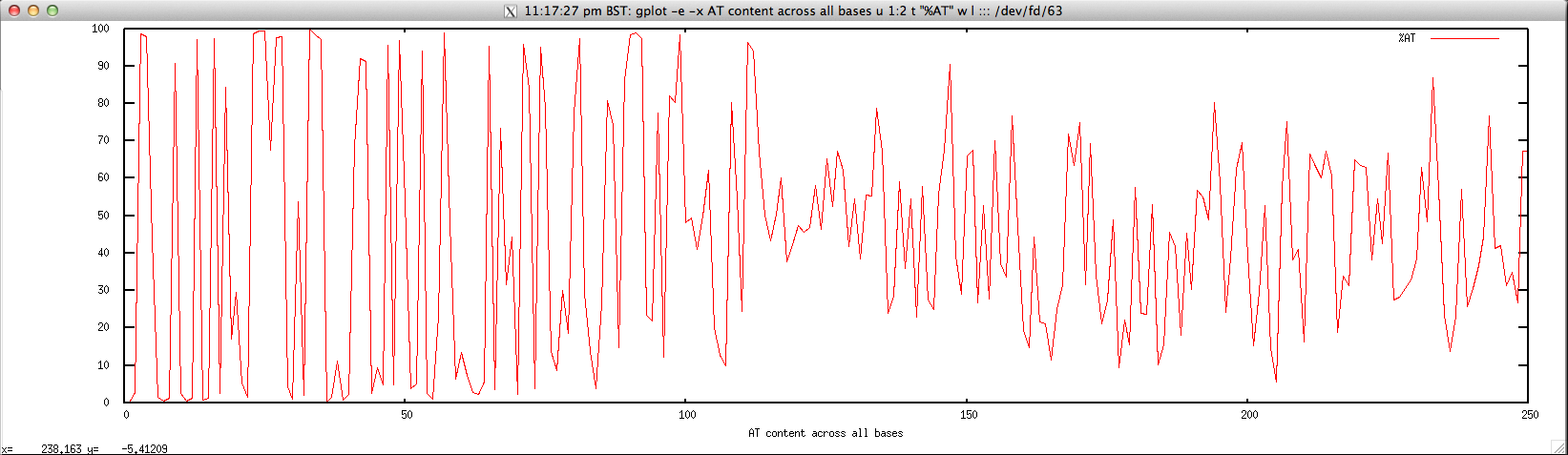
 Usage: ./perbase_ATcontent_FASTQ.sh file.fastq
Usage: ./perbase_ATcontent_FASTQ.sh file.fastq
#!/bin/bash
gplot -e -x "AT content across all bases" u 1:2 t \"%AT\" w l ::: <(cat $1 | \
perl -lne 'push @a, $_; @a = @a[@a-4..$#a]; if ($. % 4 == 0){
chomp($a[1]);
$max_j=0;
$j=0;++$i;
map{$r{$i.":".++$j}=$_}split("",$a[1]);
if($j>$max_j){
$max_j=$j;}}
}{
for($jj=1;$jj<=$max_j;$jj++){
@m=();
for($ii=1;$ii<=$i;$ii++){
unless(not defined $r{$ii.":".$jj}){
push @m,$r{$ii.":".$jj}}}
$s=join("",@m);
$l=($s=~y===c);
print ($jj."\t".((($s=~tr/AT//)/$l)*100))}')
Usage: ./perbase_ATcontent_FASTA.sh file.fasta
#!/bin/bash
gplot -e -x "AT content across all bases" u 1:2 t \"%AT\" w l ::: <(cat $1 | \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -lne 'push @a, $_; @a = @a[@a-2..$#a]; if ($. % 2 == 0){
chomp($a[1]);
$max_j=0;
$j=0;++$i;
map{$r{$i.":".++$j}=$_}split("",$a[1]);
if($j>$max_j){
$max_j=$j;}}
}{
for($jj=1;$jj<=$max_j;$jj++){
@m=();
for($ii=1;$ii<=$i;$ii++){
unless(not defined $r{$ii.":".$jj}){
push @m,$r{$ii.":".$jj}}}
$s=join("",@m);
$l=($s=~y===c);
print ($jj."\t".((($s=~tr/AT//)/$l)*100))}')
Output: ./perbase_ATcontent_FASTQ.sh forward.fastq
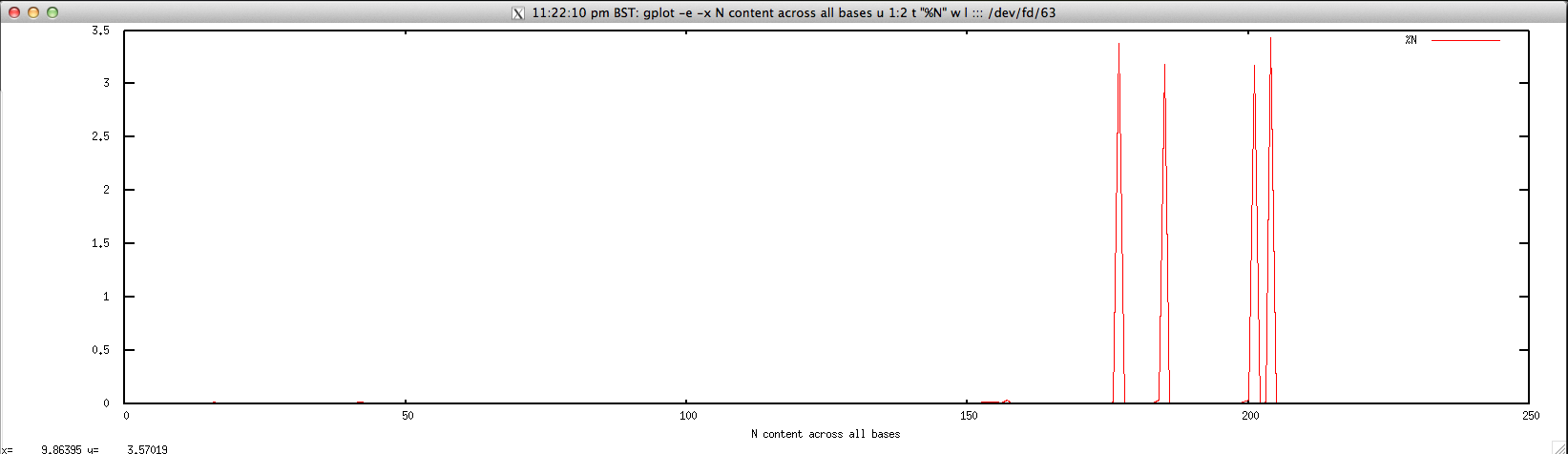
 Usage: ./perbase_Ncontent_FASTQ.sh file.fastq
Usage: ./perbase_Ncontent_FASTQ.sh file.fastq
#!/bin/bash
gplot -e -x "N content across all bases" u 1:2 t \"%N\" w l ::: <(cat $1 | \
perl -lne 'push @a, $_; @a = @a[@a-4..$#a]; if ($. % 4 == 0){
chomp($a[1]);
$max_j=0;
$j=0;
++$i;
map{$r{$i.":".++$j}=$_}split("",$a[1]);
if($j>$max_j){
$max_j=$j;}}
}{
for($jj=1;$jj<=$max_j;$jj++){
@m=();
for($ii=1;$ii<=$i;$ii++){
unless(not defined $r{$ii.":".$jj}){
push @m,$r{$ii.":".$jj}}}
$s=join("",@m);
$l=($s=~y===c);
print ($jj."\t".((($s=~tr/N//)/$l)*100))}')
Usage: ./perbase_Ncontent_FASTA.sh file.fasta
#!/bin/bash
gplot -e -x "N content across all bases" u 1:2 t \"%N\" w l ::: <(cat $1 | \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -lne 'push @a, $_; @a = @a[@a-2..$#a]; if ($. % 2 == 0){
chomp($a[1]);
$max_j=0;
$j=0;++$i;
map{$r{$i.":".++$j}=$_}split("",$a[1]);
if($j>$max_j){
$max_j=$j;}}
}{
for($jj=1;$jj<=$max_j;$jj++){
@m=();
for($ii=1;$ii<=$i;$ii++){
unless(not defined $r{$ii.":".$jj}){
push @m,$r{$ii.":".$jj}}}
$s=join("",@m);
$l=($s=~y===c);
print ($jj."\t".((($s=~tr/N//)/$l)*100))}')
Output: ./perbase_Ncontent_FASTQ.sh forward.fastq
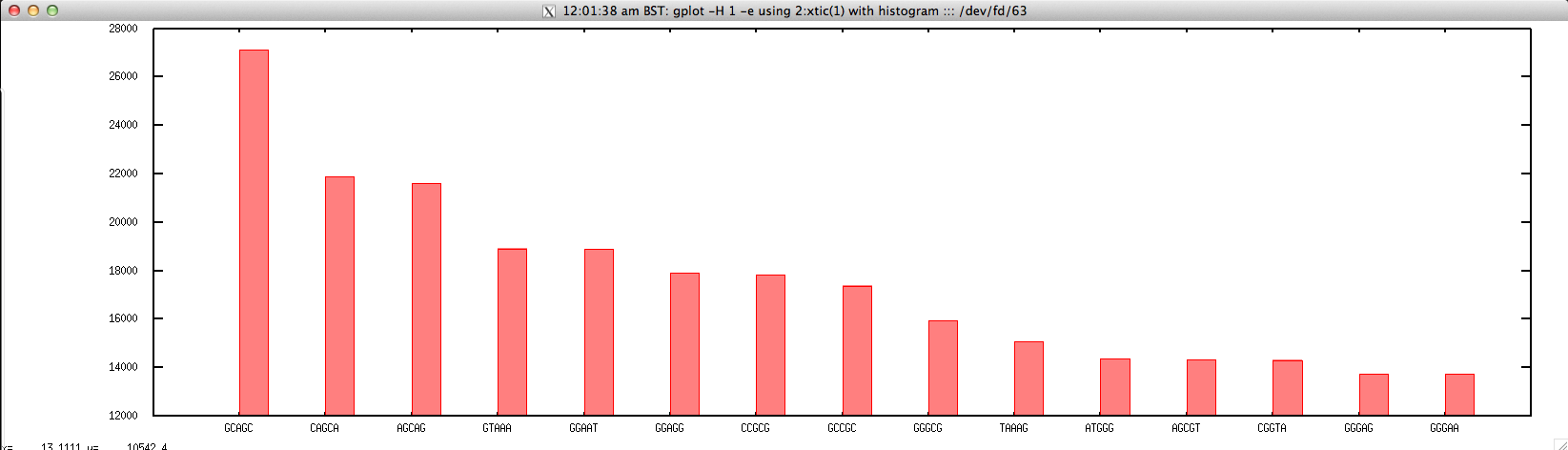
 Usage: ./top_kmer_FASTQ.sh file.fastq Kmer_size top_n
Usage: ./top_kmer_FASTQ.sh file.fastq Kmer_size top_n
#!/bin/bash
gplot -H 1 -e using '2:xtic(1)' with histogram ::: <(wordcount $1 -wordsize=$2 -stdout -auto | head -$3)
Usage: ./top_kmer_FASTA.sh file.fasta Kmer_size top_n
#!/bin/bash
gplot -H 1 -e using '2:xtic(1)' with histogram ::: <(wordcount $1 -wordsize=$2 -stdout -auto | head -$3)
Output: ./top_kmer_FASTQ.sh forward.fastq 5 15
Last Updated by Dr Umer Zeeshan Ijaz on 23/04/2014.