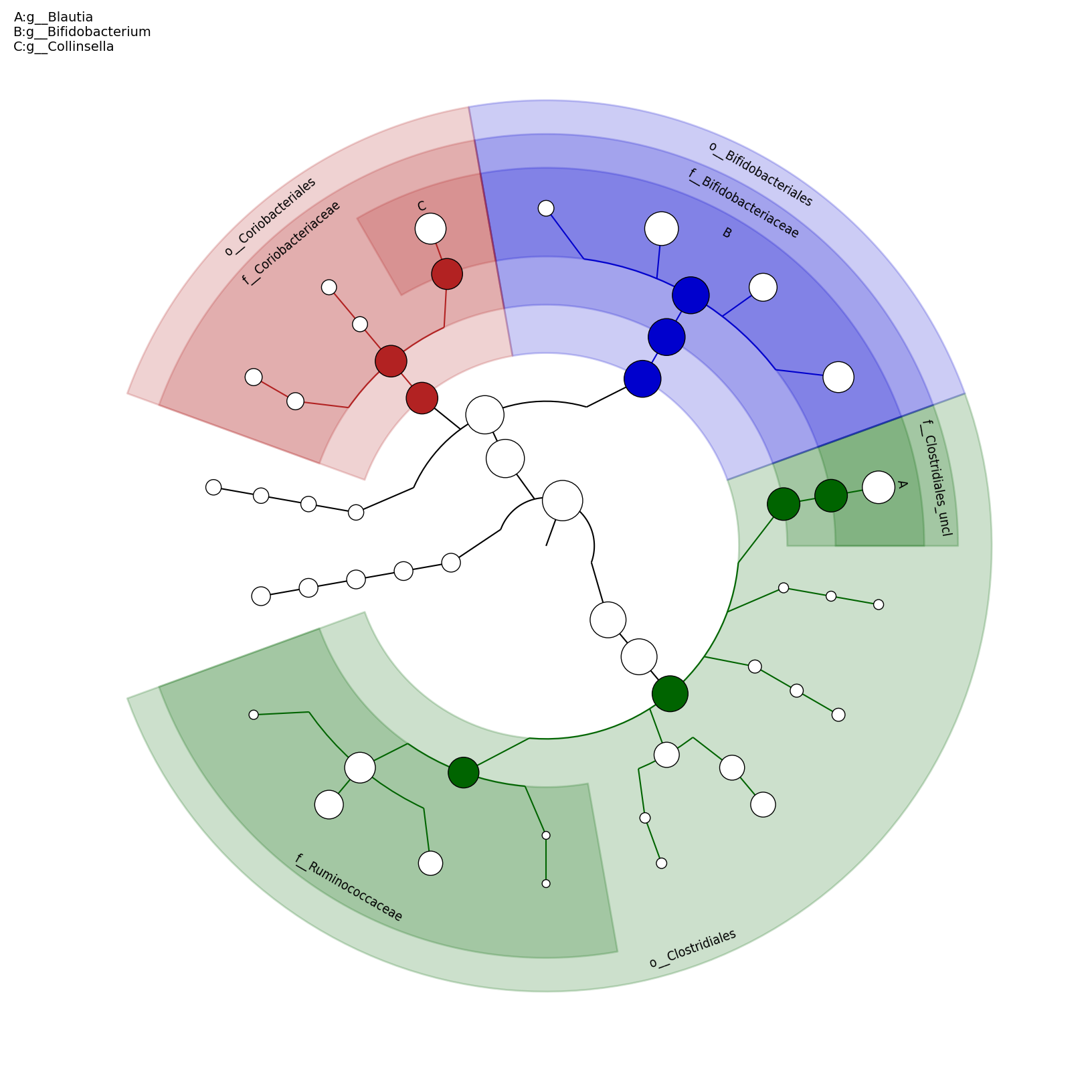
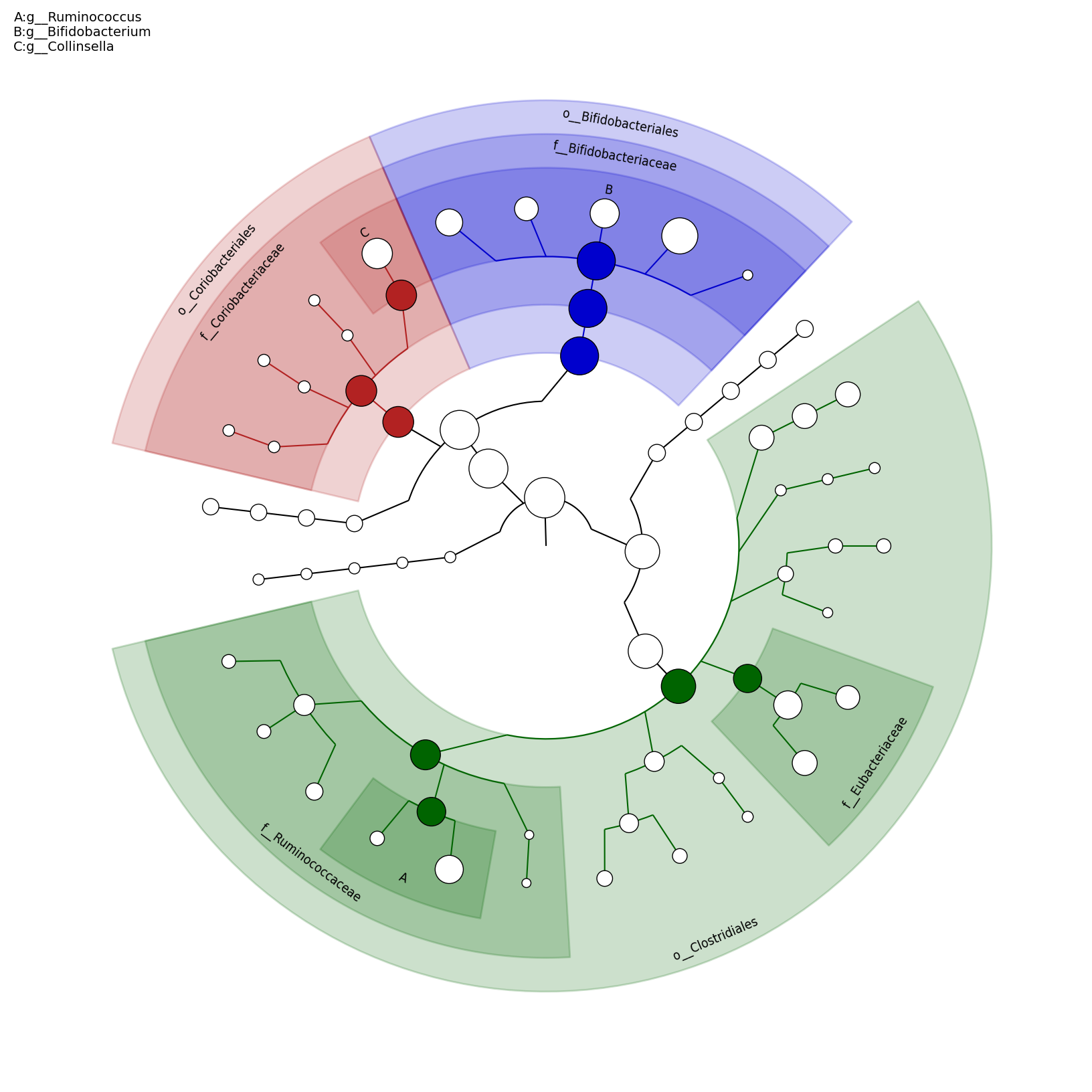
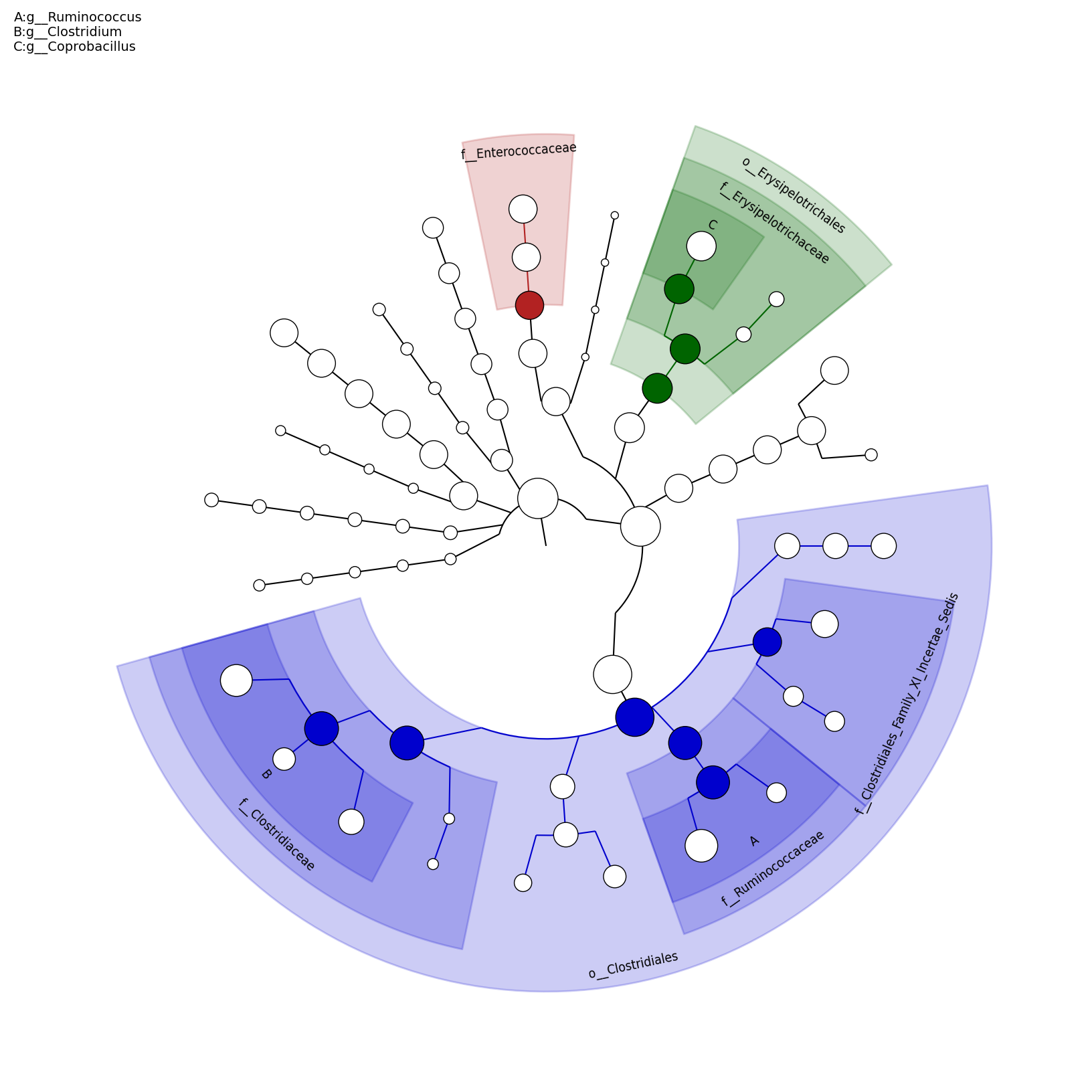
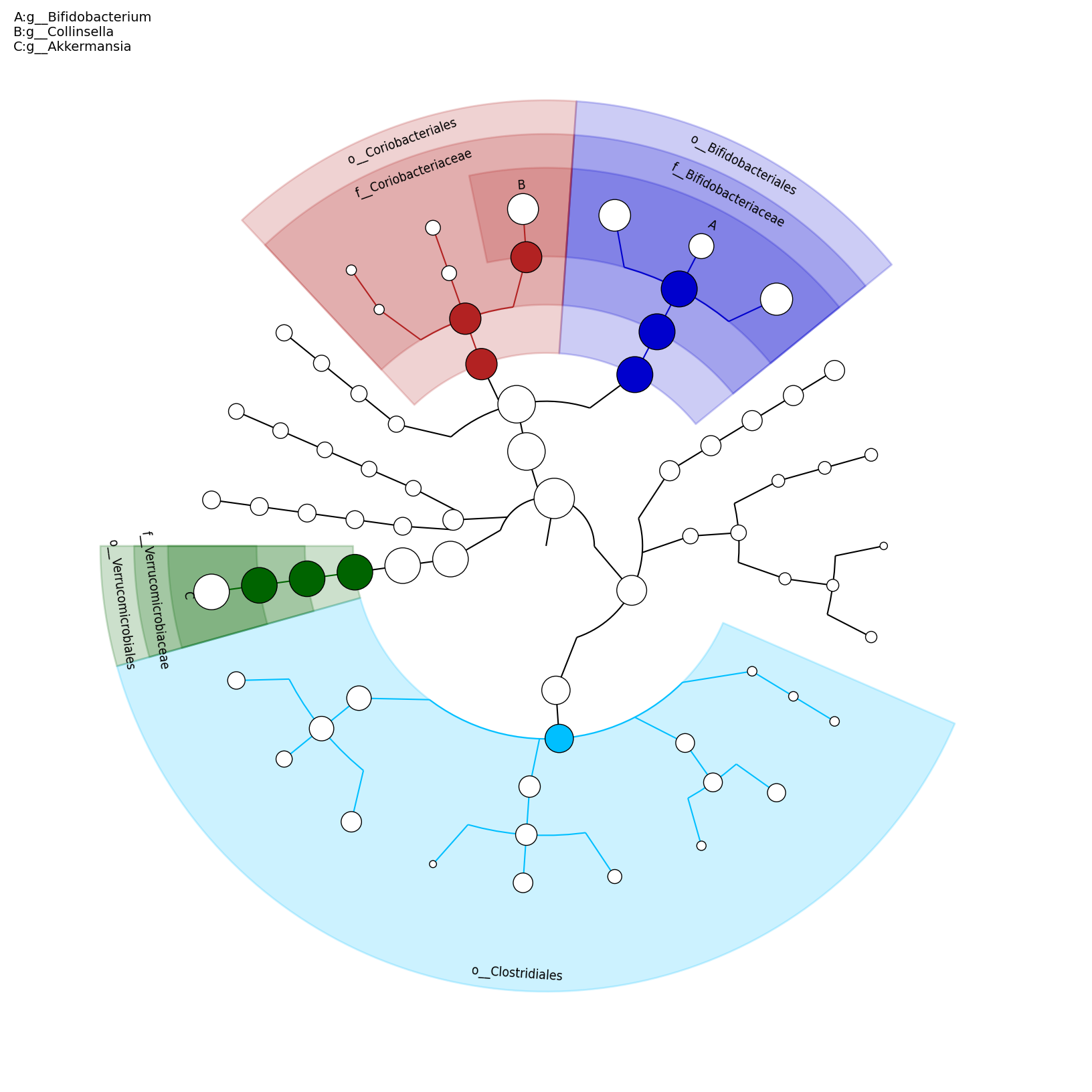
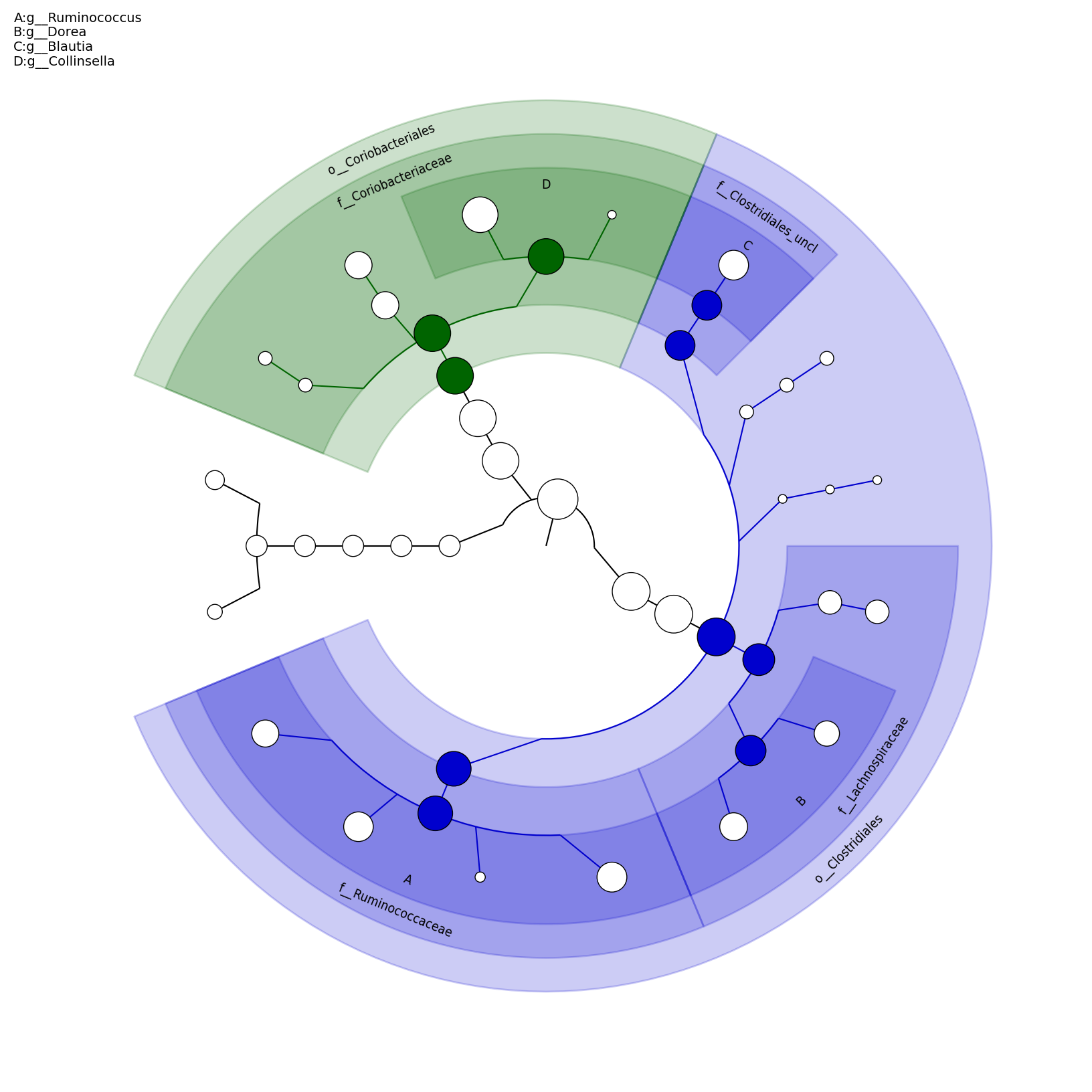
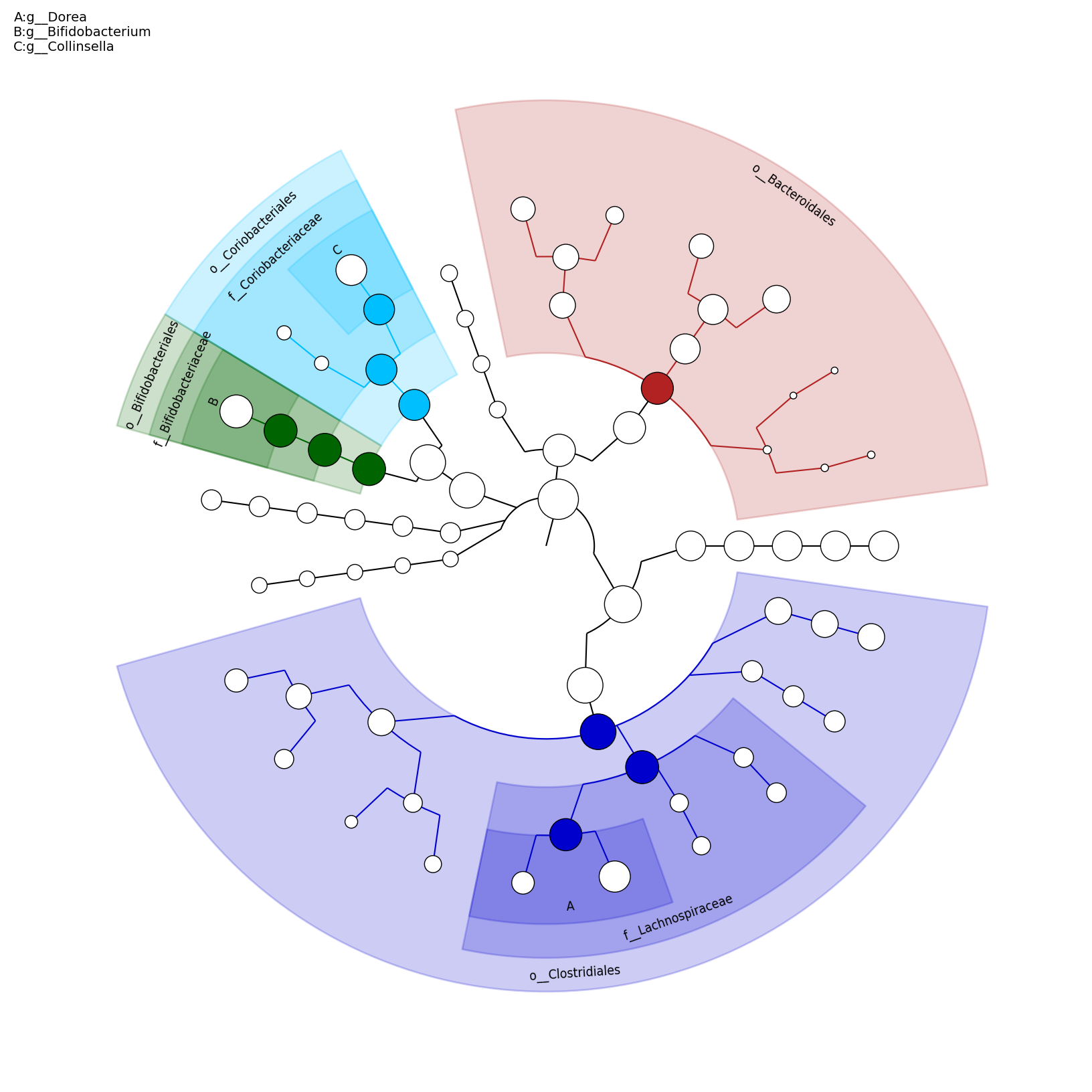
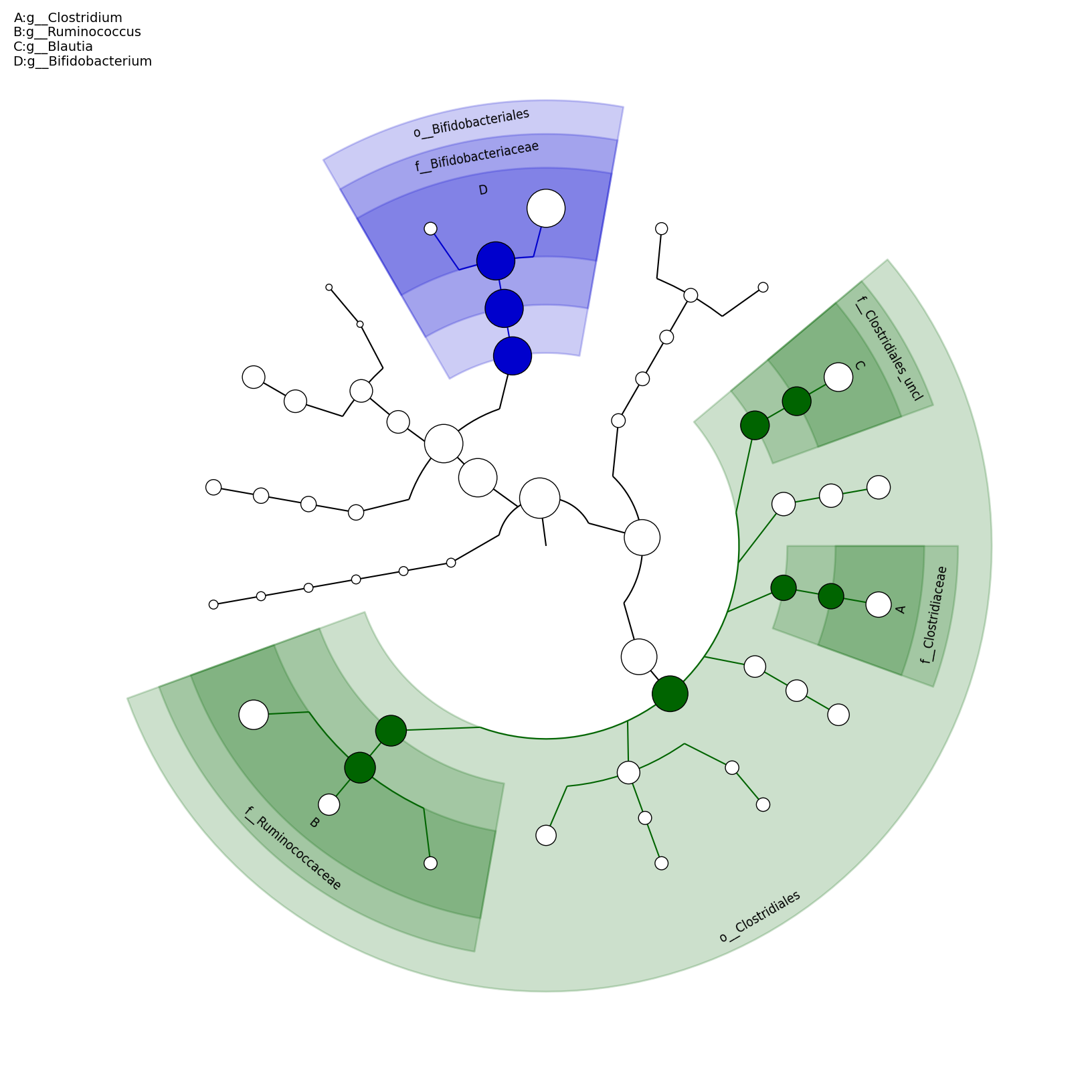
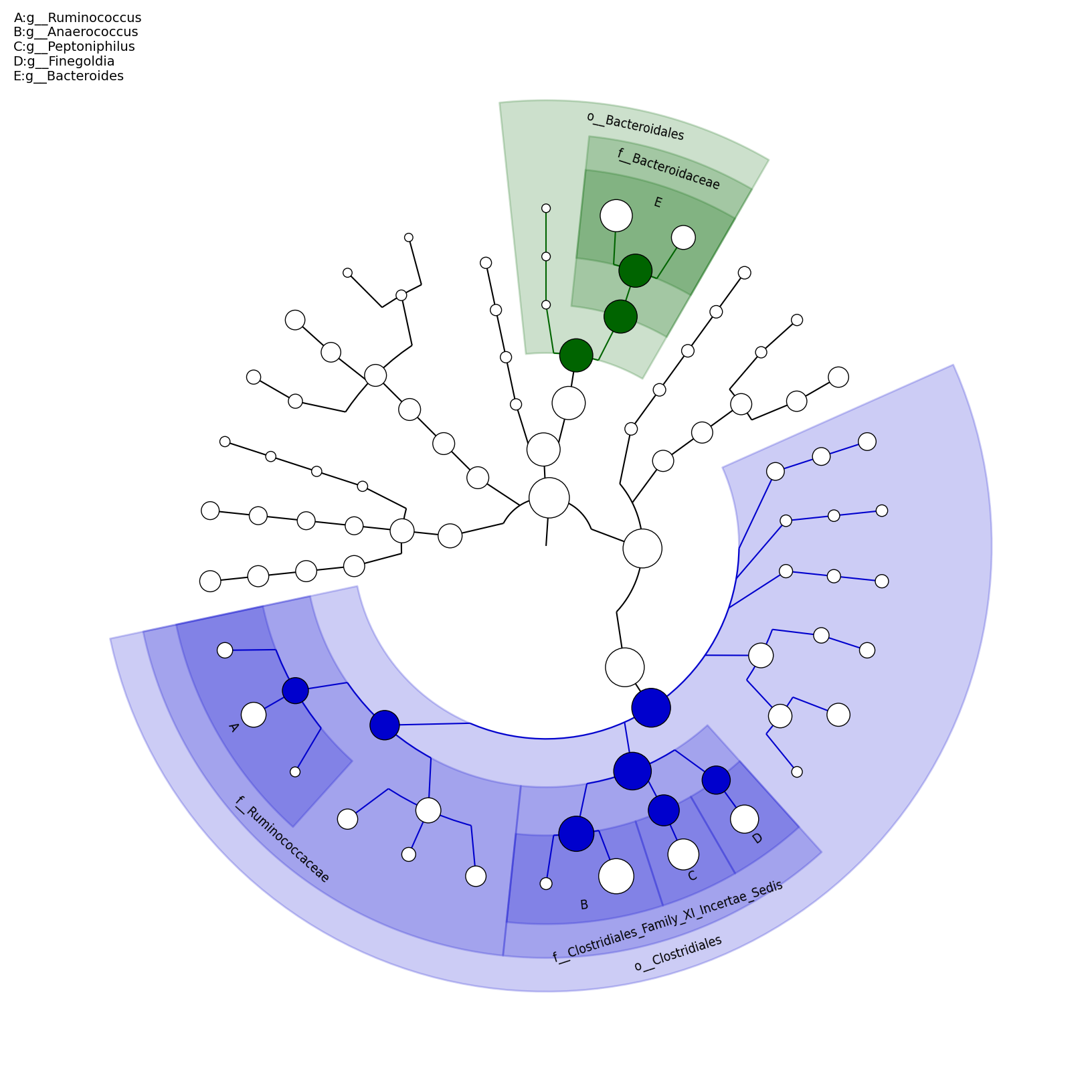
ssh -Y -p 22567 MScBioinf@quince-srv2.eng.gla.ac.ukWhen you have successfully logged on, create a folder MTutorial_YourName, go inside it and do all your processing there!
[MScBioinf@quince-srv2 ~]$ mkdir MTutorial_Umer [MScBioinf@quince-srv2 ~]$ cd MTutorial_Umer/ [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ ls [MScBioinf@quince-srv2 ~/MTutorial_Umer]$You can find the required files in the folder /home/opt/Mtutorial/Raw/. These files have been preprocessed to ensure high read quality, and have the names [C/H]ID_R[1/2].fasta. You can see that the total number of reads in *_R1.fasta matches the total number of reads in *_R2.fasta for each sample.
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ grep -c ">" /home/opt/Mtutorial/Raw/*.fasta /home/opt/Mtutorial/Raw/C13_R1.fasta:6470505 /home/opt/Mtutorial/Raw/C13_R2.fasta:6470505 /home/opt/Mtutorial/Raw/C1_R1.fasta:2751935 /home/opt/Mtutorial/Raw/C1_R2.fasta:2751935 /home/opt/Mtutorial/Raw/C27_R1.fasta:4032308 /home/opt/Mtutorial/Raw/C27_R2.fasta:4032308 /home/opt/Mtutorial/Raw/C32_R1.fasta:2071894 /home/opt/Mtutorial/Raw/C32_R2.fasta:2071894 /home/opt/Mtutorial/Raw/C45_R1.fasta:2492282 /home/opt/Mtutorial/Raw/C45_R2.fasta:2492282 /home/opt/Mtutorial/Raw/C51_R1.fasta:1363261 /home/opt/Mtutorial/Raw/C51_R2.fasta:1363261 /home/opt/Mtutorial/Raw/C56_R1.fasta:5751417 /home/opt/Mtutorial/Raw/C56_R2.fasta:5751417 /home/opt/Mtutorial/Raw/C62_R1.fasta:3581667 /home/opt/Mtutorial/Raw/C62_R2.fasta:3581667 /home/opt/Mtutorial/Raw/C68_R1.fasta:1227785 /home/opt/Mtutorial/Raw/C68_R2.fasta:1227785 /home/opt/Mtutorial/Raw/C80_R1.fasta:6168464 /home/opt/Mtutorial/Raw/C80_R2.fasta:6168464 /home/opt/Mtutorial/Raw/H117_R1.fasta:3839459 /home/opt/Mtutorial/Raw/H117_R2.fasta:3839459 /home/opt/Mtutorial/Raw/H119_R1.fasta:4965592 /home/opt/Mtutorial/Raw/H119_R2.fasta:4965592 /home/opt/Mtutorial/Raw/H128_R1.fasta:2798649 /home/opt/Mtutorial/Raw/H128_R2.fasta:2798649 /home/opt/Mtutorial/Raw/H130_R1.fasta:3757735 /home/opt/Mtutorial/Raw/H130_R2.fasta:3757735 /home/opt/Mtutorial/Raw/H132_R1.fasta:5028567 /home/opt/Mtutorial/Raw/H132_R2.fasta:5028567 /home/opt/Mtutorial/Raw/H134_R1.fasta:5432398 /home/opt/Mtutorial/Raw/H134_R2.fasta:5432398 /home/opt/Mtutorial/Raw/H139_R1.fasta:7358644 /home/opt/Mtutorial/Raw/H139_R2.fasta:7358644 /home/opt/Mtutorial/Raw/H140_R1.fasta:2836052 /home/opt/Mtutorial/Raw/H140_R2.fasta:2836052 /home/opt/Mtutorial/Raw/H142_R1.fasta:3957303 /home/opt/Mtutorial/Raw/H142_R2.fasta:3957303 /home/opt/Mtutorial/Raw/H144_R1.fasta:5096369 /home/opt/Mtutorial/Raw/H144_R2.fasta:5096369 [MScBioinf@quince-srv2 ~/MTutorial_Umer]$Reported bug: In some cases, the bash shell shifts to Python 2.7.3 from Python 2.6.6 automatically and some instructions wont run citing library not found error. A workaround is to start a new bash shell, reset $PYTHONPATH, and set the directories again:
bash
export $PYTHONPATH=''
export metaphlan_dir=/home/opt/metaphlan;export graphlan_dir=/home/opt/graphlan;export lefse_dir=/home/opt/lefse
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ mkdir subsampled_s100000 [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ cd subsampled_s100000 [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$If we want to get unique sample IDs from the file names, we can use the following bash one-liner:
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ for i in $(ls /home/opt/Mtutorial/Raw/*); do echo $(echo $i | sed 's/\(.*\)_R[12]\.fasta/\1/'); done | sort |uniq /home/opt/Mtutorial/Raw/C1 /home/opt/Mtutorial/Raw/C13 /home/opt/Mtutorial/Raw/C27 /home/opt/Mtutorial/Raw/C32 /home/opt/Mtutorial/Raw/C45 /home/opt/Mtutorial/Raw/C51 /home/opt/Mtutorial/Raw/C56 /home/opt/Mtutorial/Raw/C62 /home/opt/Mtutorial/Raw/C68 /home/opt/Mtutorial/Raw/C80 /home/opt/Mtutorial/Raw/H117 /home/opt/Mtutorial/Raw/H119 /home/opt/Mtutorial/Raw/H128 /home/opt/Mtutorial/Raw/H130 /home/opt/Mtutorial/Raw/H132 /home/opt/Mtutorial/Raw/H134 /home/opt/Mtutorial/Raw/H139 /home/opt/Mtutorial/Raw/H140 /home/opt/Mtutorial/Raw/H142 /home/opt/Mtutorial/Raw/H144 [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$In the above command, we are using regular expressions in sed to recover the IDs, for example, for a given filename, sed returns the output as:
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ echo "/home/opt/Mtutorial/Raw/H140_R2.fasta" | sed 's/\(.*\)_R[12]\.fasta/\1/' /home/opt/Mtutorial/Raw/H140 [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$If we enclose the previous for loop in $(), we can use it as a variable and nest it inside another for loop in bash. We can then use /home/opt/Mtutorial/bin/subsample_pe_fasta.sh (description given below) script to generate the subsampled files in the current folder, which will have the same names but will have 100000 reads. I have put echo in the command below to see the format of the instructions that will run in each iteration of the for loop:
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ for i in $(for i in $(ls /home/opt/Mtutorial/Raw/*); do echo $(echo $i | sed 's/\(.*\)_R[12]\.fasta/\1/'); done | sort | uniq); do echo /home/opt/Mtutorial/bin/subsample_pe_fasta.sh ${i}_R1.fasta ${i}_R2.fasta $(basename ${i})_R1.fasta $(basename ${i})_R2.fasta 100000; done /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/C1_R1.fasta /home/opt/Mtutorial/Raw/C1_R2.fasta C1_R1.fasta C1_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/C13_R1.fasta /home/opt/Mtutorial/Raw/C13_R2.fasta C13_R1.fasta C13_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/C27_R1.fasta /home/opt/Mtutorial/Raw/C27_R2.fasta C27_R1.fasta C27_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/C32_R1.fasta /home/opt/Mtutorial/Raw/C32_R2.fasta C32_R1.fasta C32_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/C45_R1.fasta /home/opt/Mtutorial/Raw/C45_R2.fasta C45_R1.fasta C45_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/C51_R1.fasta /home/opt/Mtutorial/Raw/C51_R2.fasta C51_R1.fasta C51_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/C56_R1.fasta /home/opt/Mtutorial/Raw/C56_R2.fasta C56_R1.fasta C56_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/C62_R1.fasta /home/opt/Mtutorial/Raw/C62_R2.fasta C62_R1.fasta C62_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/C68_R1.fasta /home/opt/Mtutorial/Raw/C68_R2.fasta C68_R1.fasta C68_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/C80_R1.fasta /home/opt/Mtutorial/Raw/C80_R2.fasta C80_R1.fasta C80_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/H117_R1.fasta /home/opt/Mtutorial/Raw/H117_R2.fasta H117_R1.fasta H117_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/H119_R1.fasta /home/opt/Mtutorial/Raw/H119_R2.fasta H119_R1.fasta H119_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/H128_R1.fasta /home/opt/Mtutorial/Raw/H128_R2.fasta H128_R1.fasta H128_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/H130_R1.fasta /home/opt/Mtutorial/Raw/H130_R2.fasta H130_R1.fasta H130_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/H132_R1.fasta /home/opt/Mtutorial/Raw/H132_R2.fasta H132_R1.fasta H132_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/H134_R1.fasta /home/opt/Mtutorial/Raw/H134_R2.fasta H134_R1.fasta H134_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/H139_R1.fasta /home/opt/Mtutorial/Raw/H139_R2.fasta H139_R1.fasta H139_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/H140_R1.fasta /home/opt/Mtutorial/Raw/H140_R2.fasta H140_R1.fasta H140_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/H142_R1.fasta /home/opt/Mtutorial/Raw/H142_R2.fasta H142_R1.fasta H142_R2.fasta 100000 /home/opt/Mtutorial/bin/subsample_pe_fasta.sh /home/opt/Mtutorial/Raw/H144_R1.fasta /home/opt/Mtutorial/Raw/H144_R2.fasta H144_R1.fasta H144_R2.fasta 100000 [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$
Store first s elements into R. for each element in position k = s+1 to n , accept it with probability s/k if accepted, choose a random element from R to replace.Using reservoir sampling, we have written several bash scripts that allow us to subsample single-end and paired-end fasta as well as fastq files. The content of these scripts are as follows:
Example usage: ./subsample_se_fasta.sh s.fasta s_sub.fasta 100000
#!/bin/bash
awk '/^>/ {printf("\n%s\n",$0);next; } { printf("%s",$0);} END {printf("\n");}' < $1 | awk 'NR>1{ printf("%s",$0); n++; if(n%2==0) { printf("\n");} else { printf("\t");} }' | awk -v k=$3 'BEGIN{srand(systime() + PROCINFO["pid"]);}{s=x++<k?x-1:int(rand()*x);if(s<k)R[s]=$0}END{for(i in R)print R[i]}' | awk -F"\t" -v f="$2" '{print $1"\n"$2 > f}'
Example usage: ./subsample_se_fastq.sh s.fastq s_sub.fastq 100000
#!/bin/bash
cat $1 | awk '{ printf("%s",$0); n++; if(n%4==0) { printf("\n");} else { printf("\t");} }' | awk -v k=$3 'BEGIN{srand(systime() + PROCINFO["pid"]);}{s=x++<k?x-1:int(rand()*x);if(s<<)R[s]=$0}END{for(i in R)print R[i]}' | awk -F"\t" -v f="$2" '{print $1"\n"$2"\n"$3"\n"$4 > f}'
Example usage: ./subsample_pe_fasta.sh f.fasta r.fasta f_sub.fasta r_sub.fasta 100000
#!/bin/bash
paste <(awk '/^>/ {printf("\n%s\n",$0);next; } {printf("%s",$0);} END {printf("\n");}' < $1) <(awk '/^>/ {printf("\n%s\n",$0);next; } { printf("%s",$0);} END {printf("\n");}' < $2) | awk 'NR>1{ printf("%s",$0); n++; if(n%2==0) { printf("\n");} else { printf("\t");} }' | awk -v k=$5 'BEGIN{srand(systime() + PROCINFO["pid"]);}{s=x++<k?x-1:int(rand()*x);if(s<k)R[s]=$0}END{for(i in R)print R[i]}' | awk -F"\t" -v f="$3" -v r="$4" '{print $1"\n"$3 > f;print $2"\n"$4 > r}'
Example usage: ./subsample_pe_fastq.sh f.fastq r.fastq f_sub.fastq r_sub.fastq 100000
#!/bin/bash
paste $1 $2 | awk '{ printf("%s",$0); n++; if(n%4==0) { printf("\n");} else { printf("\t");} }' | awk -v k=$5 'BEGIN{srand(systime() + PROCINFO["pid"]);}{s=x++<<k?x-1:int(rand()*x);if(s<k)R[s]=$0}END{for(i in R)print R[i]}' | awk -F"\t" -v f="$3" -v r="$4" '{print $1"\n"$3"\n"$5"\n"$7 > f;print $2"\n"$4"\n"$6"\n"$8 > r}'
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ for i in $(for i in $(ls /home/opt/Mtutorial/Raw/*); do echo $(echo $i | sed 's/\(.*\)_R[12]\.fasta/\1/'); done | sort | uniq); do /home/opt/Mtutorial/bin/subsample_pe_fasta.sh ${i}_R1.fasta ${i}_R2.fasta $(basename ${i})_R1.fasta $(basename ${i})_R2.fasta 100000; done
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ for i in $(ls /home/opt/Mtutorial/Raw/*); do echo $(echo $i | sed 's/\(.*\)_R[12]\.fasta/\1/'); done | sort | uniq | parallel -k '/home/opt/Mtutorial/bin/subsample_pe_fasta.sh {}_R1.fasta {}_R2.fasta $(basename {})_R1.fasta $(basename {})_R2.fasta 100000'
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ grep -c ">" *.fasta C13_R1.fasta:100000 C13_R2.fasta:100000 C1_R1.fasta:100000 C1_R2.fasta:100000 C27_R1.fasta:100000 C27_R2.fasta:100000 C32_R1.fasta:100000 C32_R2.fasta:100000 C45_R1.fasta:100000 C45_R2.fasta:100000 C51_R1.fasta:100000 C51_R2.fasta:100000 C56_R1.fasta:100000 C56_R2.fasta:100000 C62_R1.fasta:100000 C62_R2.fasta:100000 C68_R1.fasta:100000 C68_R2.fasta:100000 C80_R1.fasta:100000 C80_R2.fasta:100000 H117_R1.fasta:100000 H117_R2.fasta:100000 H119_R1.fasta:100000 H119_R2.fasta:100000 H128_R1.fasta:100000 H128_R2.fasta:100000 H130_R1.fasta:100000 H130_R2.fasta:100000 H132_R1.fasta:100000 H132_R2.fasta:100000 H134_R1.fasta:100000 H134_R2.fasta:100000 H139_R1.fasta:100000 H139_R2.fasta:100000 H140_R1.fasta:100000 H140_R2.fasta:100000 H142_R1.fasta:100000 H142_R2.fasta:100000 H144_R1.fasta:100000 H144_R2.fasta:100000 [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$We can check that we are not losing the paired-end information and have the same IDs for the first two forward reads in *_R1.fasta files that we have in *_R2.fasta files:
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ head -4 C13_*.fasta ==> C13_R1.fasta <== >D00255:65:H74RMADXX:1:2201:6972:15437/1 TGACAGATGGGCACAGGCAGAAGCCGTTCCTTTTTGGATTCCTGCTGTATCAGAACATGCTGTGGTTTGTGAGTGATGATGCAAAACTGCCGAACATGGTGGAACAGATTCGGGAAGAACTATATGAGGGGTTCTCTATTCAGGATTTTCT >D00255:63:H74RLADXX:1:1215:14403:77265/1 GTGTCGATGCAGTAGGCCATGCGGAAGTACCCGGACACGCCGAAGCTGTCGGACGGCACCAGGATCAGGTCGTATTCCTTGGCCTTCATGCAGAAGGCGTTCGCG ==> C13_R2.fasta <== >D00255:65:H74RMADXX:1:2201:6972:15437/2 CAGATACATATTCCAGTATAGTATCCGTGTCGCTGTGATAACTCCCTGCGGTTCTGCATGATTGCTTCATTGAGGTGGGCTTCTTTCCTGTCAAGCACTTTTTCTTCCATACGTTCCAGCCCATCTTCAATTTTCTGAAGGAAGCTCATAT >D00255:63:H74RLADXX:1:1215:14403:77265/2 TTCTACGACAACACGCTGACCTGCTACTCCTGGTCGAAGTCGCTGAGCCTGCCGGGCGAGCGCATCGGCTACGTGGCAGTCAACCCGCGGGCGACCGACGCCGAACTCATCGTGCCGATGTGCGGGCAGATCTCACGCGGCACCGGCCACA [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$Now we concatenate both *_R1.fasta and *_R2.fasta together and produce *_R12.fasta. Resulting files should have 200,000 reads:
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ for i in $(for i in $(ls *); do echo $(echo $i | sed 's/\(.*\)_R[12]\.fasta/\1/'); done | sort | uniq); do cat ${i}_R1.fasta ${i}_R2.fasta > ${i}_R12.fasta; rm ${i}_R?.fasta; done [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ ls C13_R12.fasta C27_R12.fasta C45_R12.fasta C56_R12.fasta C68_R12.fasta H117_R12.fasta H128_R12.fasta H132_R12.fasta H139_R12.fasta H142_R12.fasta C1_R12.fasta C32_R12.fasta C51_R12.fasta C62_R12.fasta C80_R12.fasta H119_R12.fasta H130_R12.fasta H134_R12.fasta H140_R12.fasta H144_R12.fasta [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ grep -c ">" *.fasta C13_R12.fasta:200000 C1_R12.fasta:200000 C27_R12.fasta:200000 C32_R12.fasta:200000 C45_R12.fasta:200000 C51_R12.fasta:200000 C56_R12.fasta:200000 C62_R12.fasta:200000 C68_R12.fasta:200000 C80_R12.fasta:200000 H117_R12.fasta:200000 H119_R12.fasta:200000 H128_R12.fasta:200000 H130_R12.fasta:200000 H132_R12.fasta:200000 H134_R12.fasta:200000 H139_R12.fasta:200000 H140_R12.fasta:200000 H142_R12.fasta:200000 H144_R12.fasta:200000 [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$The first half of *_R12.fasta consists of reads from *_R1.fasta, and the second half consists of reads from *_R2.fasta in the order they appear in the respective files:
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ awk '/^>/{++n;if(n==1 || n==100001) print}' C13_*.fasta >D00255:65:H74RMADXX:1:2201:6972:15437/1 >D00255:65:H74RMADXX:1:2201:6972:15437/2 [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$Next we arrange all the files in the current directory so that each sample file has a separate folder and the fasta file goes inside a "Raw" subfolder:
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ for i in $(awk -F"_" '{print $1}' <(ls *.fasta) | sort | uniq); do mkdir $i; mkdir $i/Raw; mv ${i}_*.fasta $i/Raw/.; done [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ ls C1 C13 C27 C32 C45 C51 C56 C62 C68 C80 H117 H119 H128 H130 H132 H134 H139 H140 H142 H144 [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ ls C1 Raw [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ ls C1/Raw C1_R12.fasta [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ export metaphlan_dir=/home/opt/metaphlan;export graphlan_dir=/home/opt/graphlan;export lefse_dir=/home/opt/lefse
The latest version of MetaPhlAn supports multifastq files using BowTie2 which is much faster than the NCBI BlastN (You can use this command instead when BlastN is the only option: metaphlan_dir/metaphlan.py Raw/*.fasta --blastdb $metaphlan_dir/blastdb/mpa --blastout ${i}.txt)
but a non-local hit policy for BowTie2 (i.e. '--bt2_ps very-sensitive') is recommended for avoiding overly-sensitive hits when using non-preprocessed fastq files.
Now run metaphlan inside each folder. Here we are using 8 cores and you may want to adjust the number of cores depending on the server workload:
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ for i in $(ls * -d); do cd $i; $metaphlan_dir/metaphlan.py Raw/*.fasta --bowtie2db $metaphlan_dir/bowtie2db/mpa --bt2_ps sensitive-local --nproc 8 > ${i}.txt ;cd ..; done
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ for i in $(ls * -d); do rm $i/*.txt; rm $i/Raw/*.txt; done
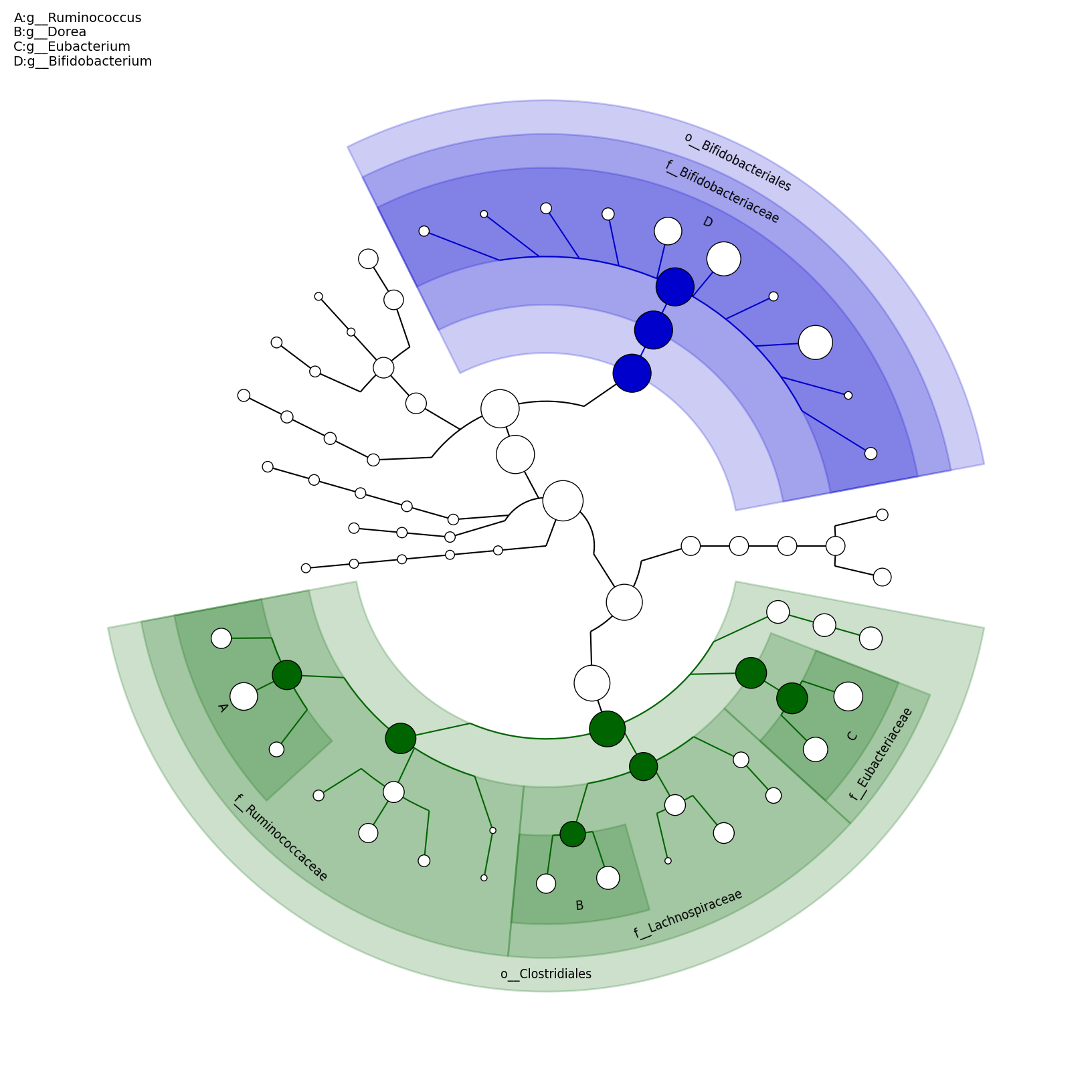
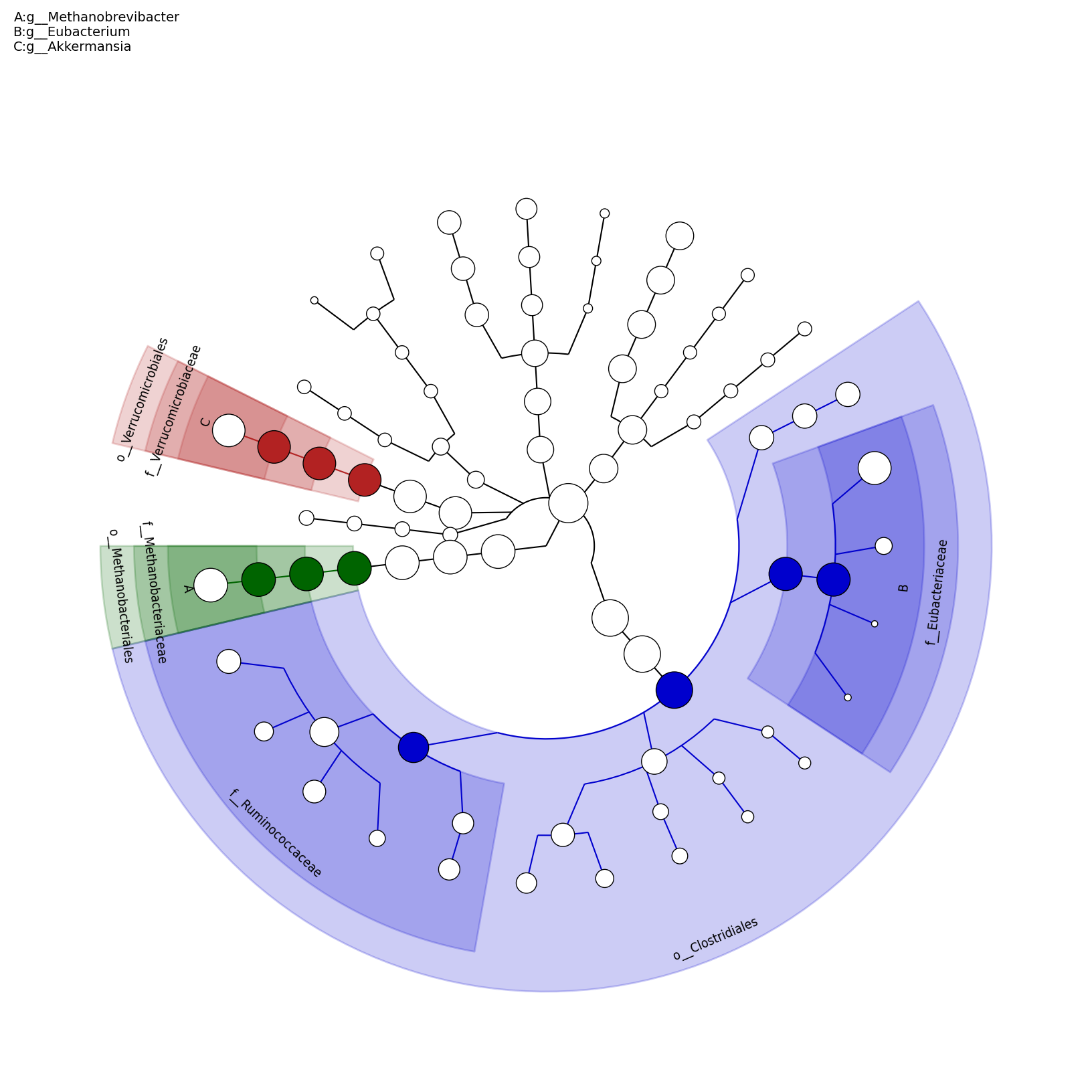
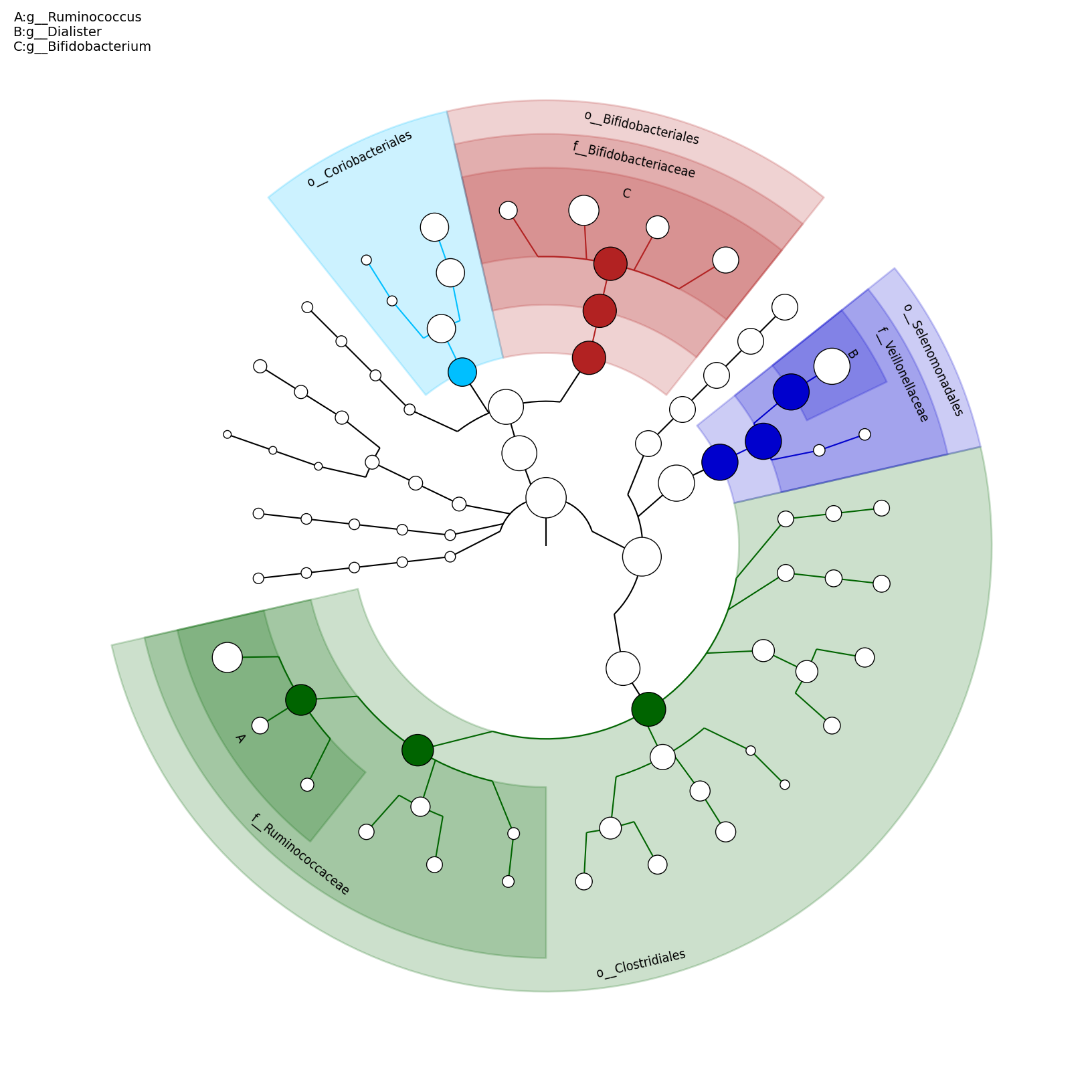
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ for i in $(ls * -d); do cd $i; $metaphlan_dir/plotting_scripts/metaphlan2graphlan.py ${i}.txt --tree_file ${i}.tree.txt --annot_file ${i}.annot.txt; $graphlan_dir/graphlan_annotate.py --annot ${i}.annot.txt ${i}.tree.txt ${i}.xml; $graphlan_dir/graphlan.py --dpi 200 ${i}.xml ${i}.png ;cd ..; done
As a result, the following png files are generated:
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ ls */*.png C13/C13.png C27/C27.png C45/C45.png C56/C56.png C68/C68.png H117/H117.png H128/H128.png H132/H132.png H139/H139.png H142/H142.png C1/C1.png C32/C32.png C51/C51.png C62/C62.png C80/C80.png H119/H119.png H130/H130.png H134/H134.png H140/H140.png H144/H144.png [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$
| C1
|
C13
|
|
C27
|
C32
|
|
C45
|
C51
|
|
C56
|
C62
|
|
C68
|
C80
|
|
H117
|
H119
|
|
H128
|
H130
|
|
H132
|
H134
|
|
H139
|
H140
|
|
H142
|
H144
|
[MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ cd .. [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ shopt -s extglob [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ $metaphlan_dir/utils/merge_metaphlan_tables.py subsampled_s100000/*/!(*.annot|*.tree).txt > merged_abundance_table.txt [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ ls merged_abundance_table.txt subsampled_s100000 [MScBioinf@quince-srv2 ~/MTutorial_Umer]$Now view the first few records of the abundance table:
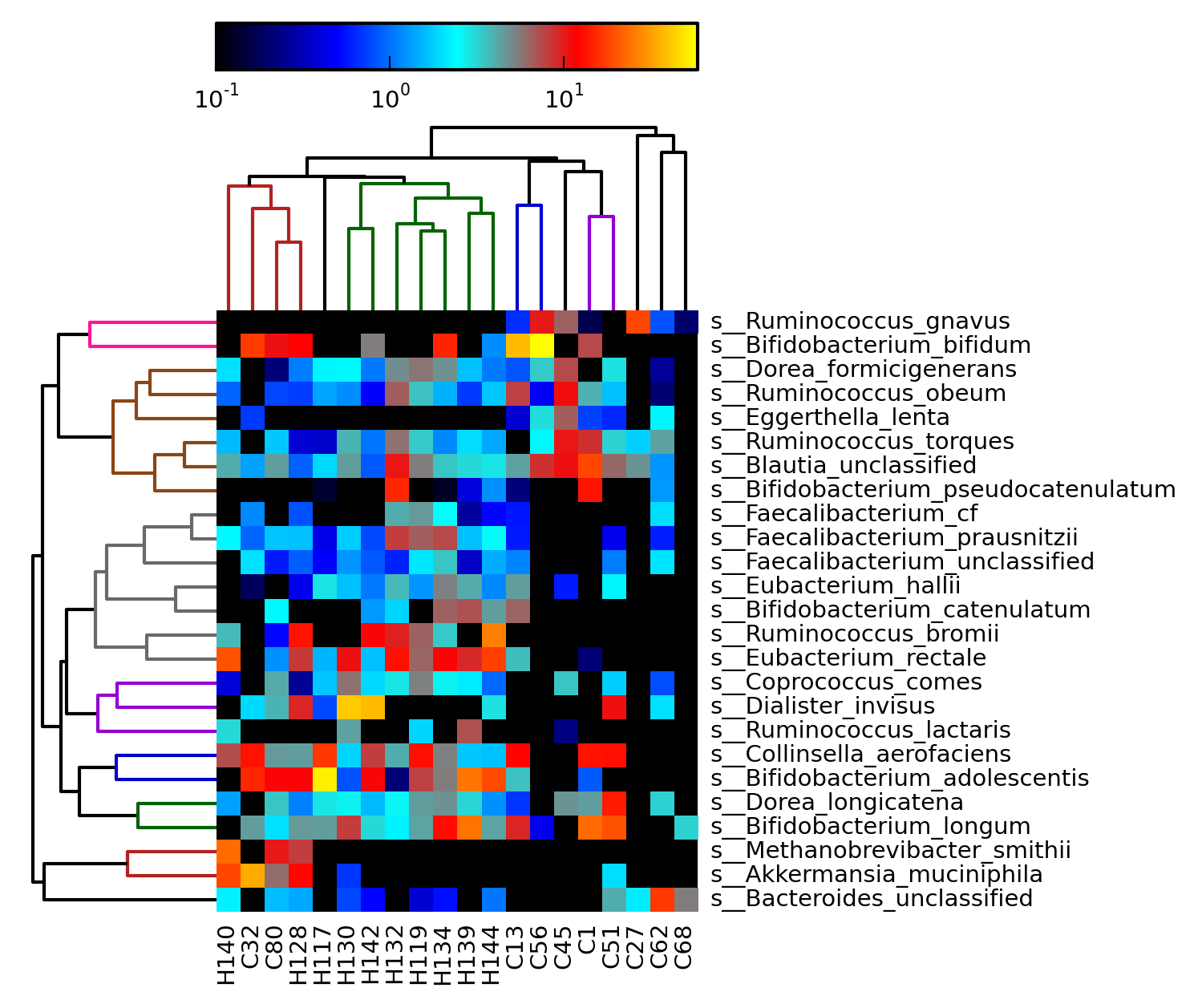
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ head merged_abundance_table.txt ID C13 C1 C27 C32 C45 C51 C56 C62 C68 C80 H117 H119 H128 H130 H132 H134 H139 H140 H142 H144 k__Archaea 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.14858 23.24301 0.0 0.0 k__Archaea|p__Euryarchaeota 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.14858 23.24301 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Halobacteria 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.14858 0.0 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Halobacteria|o__Halobacteriales 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.14858 0.0 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Halobacteria|o__Halobacteriales|f__Halobacteriales_unclassified 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.14858 0.0 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Methanobacteria 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.0 23.24301 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Methanobacteria|o__Methanobacteriales 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.0 23.24301 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Methanobacteria|o__Methanobacteriales|f__Methanobacteriaceae 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.0 23.24301 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Methanobacteria|o__Methanobacteriales|f__Methanobacteriaceae|g__Methanobrevibacter 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.0 23.24301 0.0 0.0 [MScBioinf@quince-srv2 ~/MTutorial_Umer]$Additionally, the MetaPhlAn output can now be visualized with Krona (allows interactivity) and can be converted BIOM format (specified using the --biom_output_file switch) that is used in Qiime. Next, we convert the merged_abundance_table.txt into an abundance heapmap to see if there is any clustering within datasets belonging to the same class (you can see there is):
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ $metaphlan_dir/plotting_scripts/metaphlan_hclust_heatmap.py -c bbcry --top 25 --minv 0.1 -s log --in merged_abundance_table.txt --out abundance_heatmap.png
abundance_heatmap.png
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ sed 's/\([CH]\)[0-9]*/\1/g' merged_abundance_table.txt > merged_abundance_table.2lefse.txt [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ head merged_abundance_table.2lefse.txt ID C C C C C C C C C C H H H H H H H H H H k__Archaea 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.14858 23.24301 0.0 0.0 k__Archaea|p__Euryarchaeota 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.14858 23.24301 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Halobacteria 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.14858 0.0 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Halobacteria|o__Halobacteriales 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.14858 0.0 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Halobacteria|o__Halobacteriales|f__Halobacteriales_unclassified 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.14858 0.0 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Methanobacteria 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.0 23.24301 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Methanobacteria|o__Methanobacteriales 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.0 23.24301 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Methanobacteria|o__Methanobacteriales|f__Methanobacteriaceae 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.0 23.24301 0.0 0.0 k__Archaea|p__Euryarchaeota|c__Methanobacteria|o__Methanobacteriales|f__Methanobacteriaceae|g__Methanobrevibacter 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0 10.27356 0.0 0.0 9.68621 0.0 0.0 0.0 0.0 23.24301 0.0 0.0 [MScBioinf@quince-srv2 ~/MTutorial_Umer]$However, you can run lefse only when rpy2 package is installed which only works with Python 2.7+ and not Python 2.6.6 that we used for previous analysis. Furthermore, rpy2 package complains with the older versions of R. I have compiled rpy2 package in Python 2.7.3 using a different version of R. Thus everytime, when we want to use lefse, we must copy my specially-compiled rpy2 package to the current folder, switch to python 2.7.3, use lefse commands and when we are done, we discard rpy2 folder and switch back to native Python 2.6.6. To do so, I have created two shell scripts (enable_python_lefse_rpy2.sh and disable_python_lefse_rpy2.sh) that must be run in pair in a new bash shell. After exiting from the shell, we will get back to our original settings.
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ bash [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ source /home/opt/Mtutorial/bin/enable_python2.7_lefse_rpy2.shThe first LEfSe step consists of formatting the input table, making sure the class information is in the first row and scaling the values in [0,1M] which is useful for numerical computational reasons.
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ python $lefse_dir/format_input.py merged_abundance_table.2lefse.txt merged_abundance_table.lefse -c 1 -o 1000000
Now, the LEfSe biomarker discovery tool can be used with default statistical options. Here we change one default parameter to increase the threshold on the LDA effect size from 2 (default) to 4.
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ python $lefse_dir/run_lefse.py merged_abundance_table.lefse merged_abundance_table.lefse.out -l 4
The results of the operation can now be displayed plotting the resulting list of biomarkers with corresponding effect sizes.
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ python $lefse_dir/plot_res.py --dpi 300 merged_abundance_table.lefse.out lefse_biomarkers.png
lefse_biomarkers.png
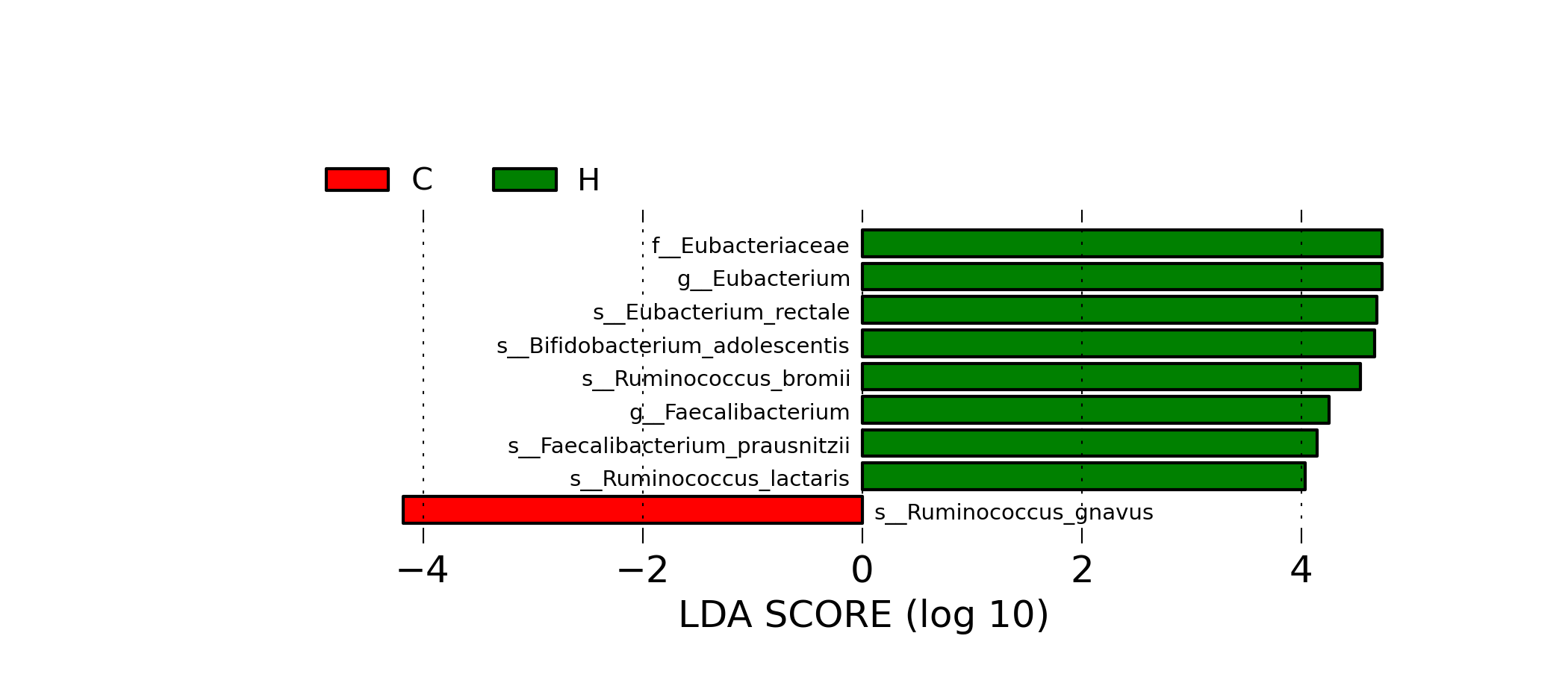
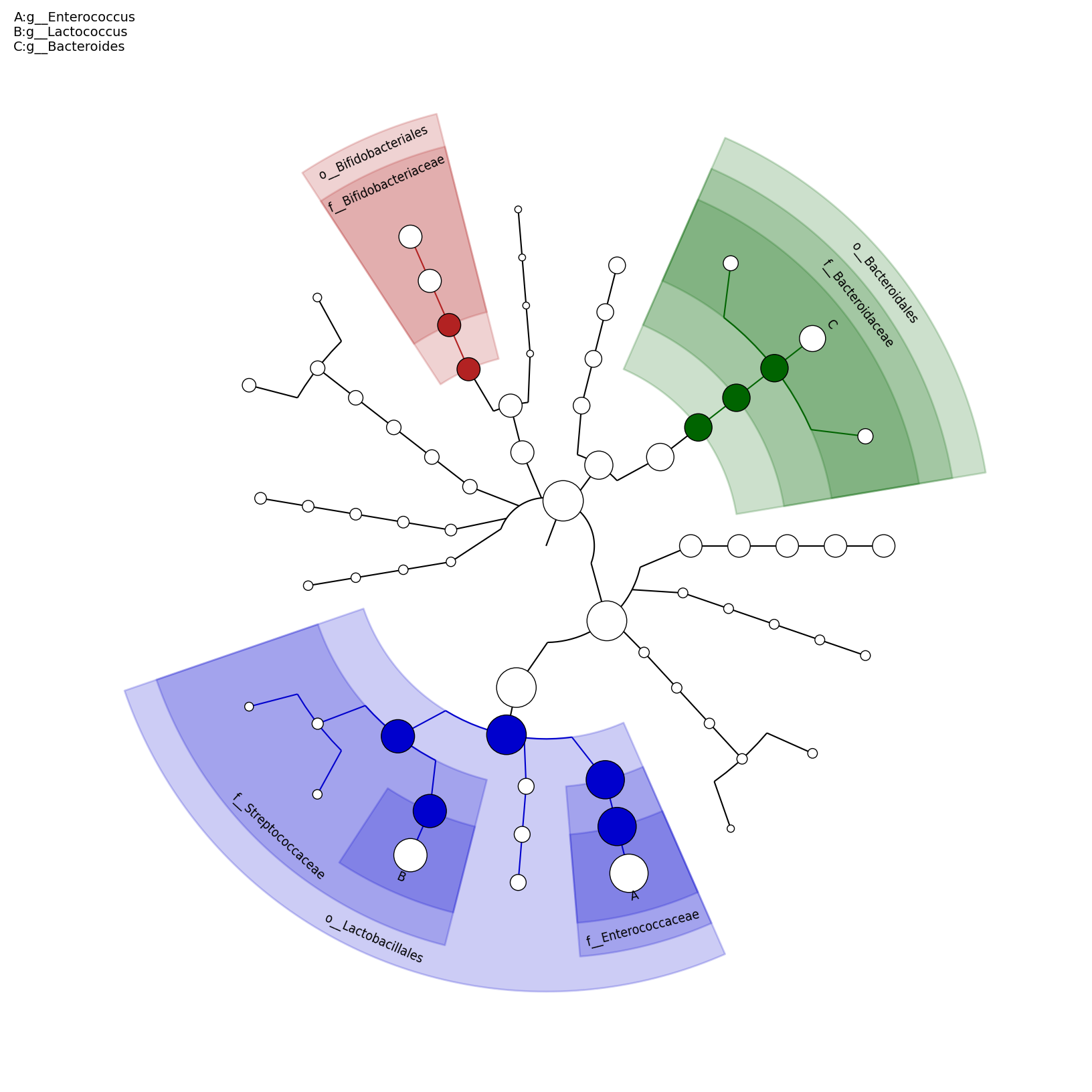
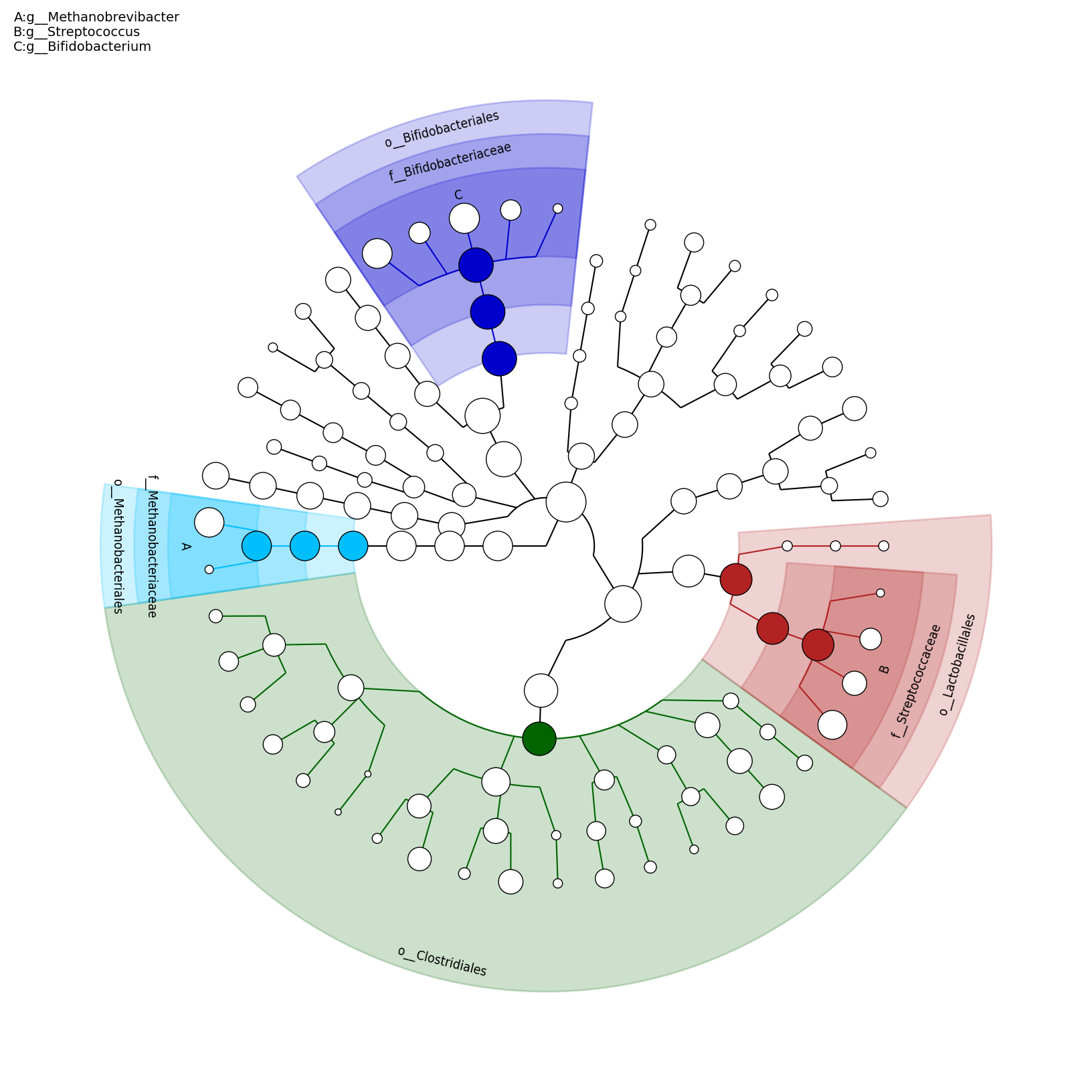
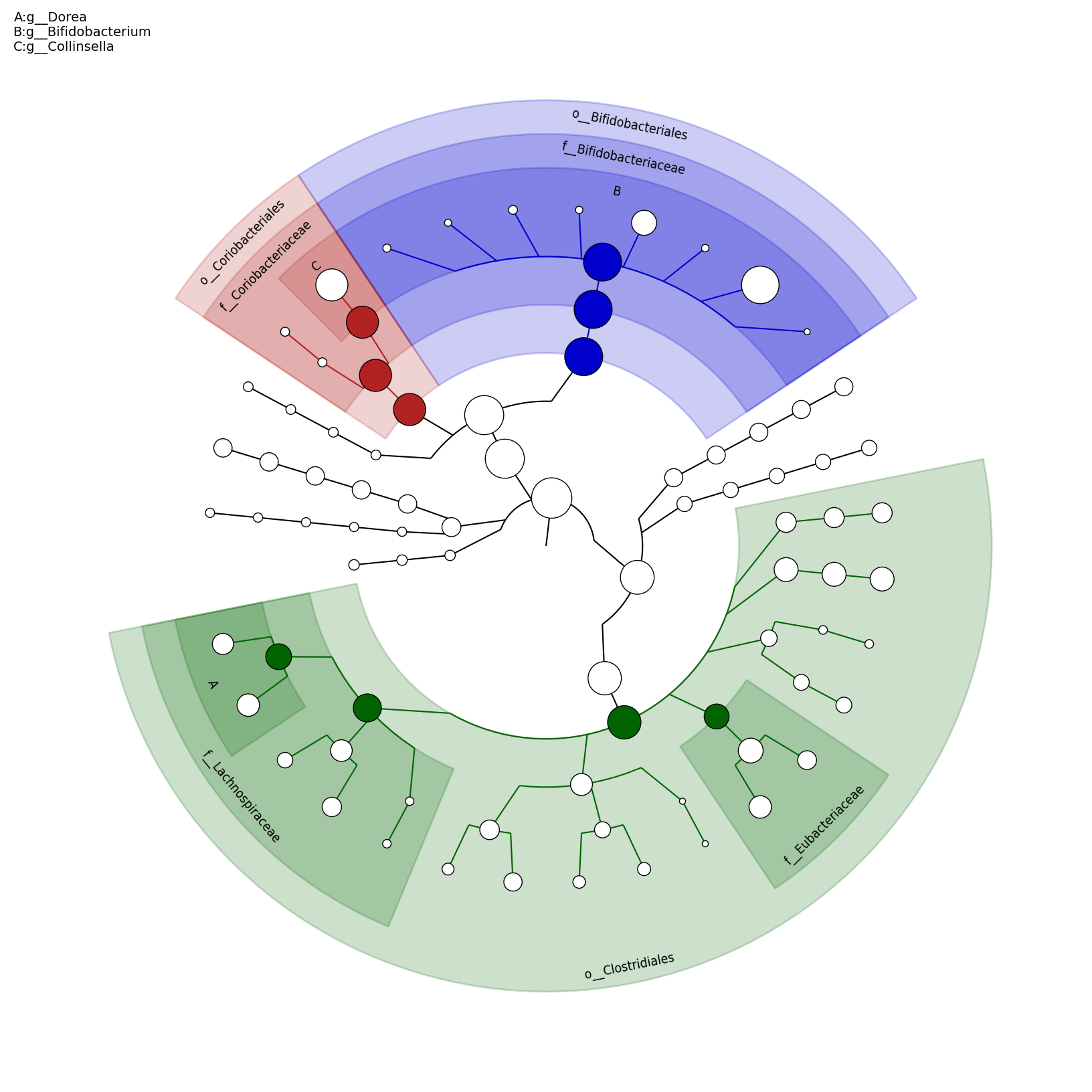
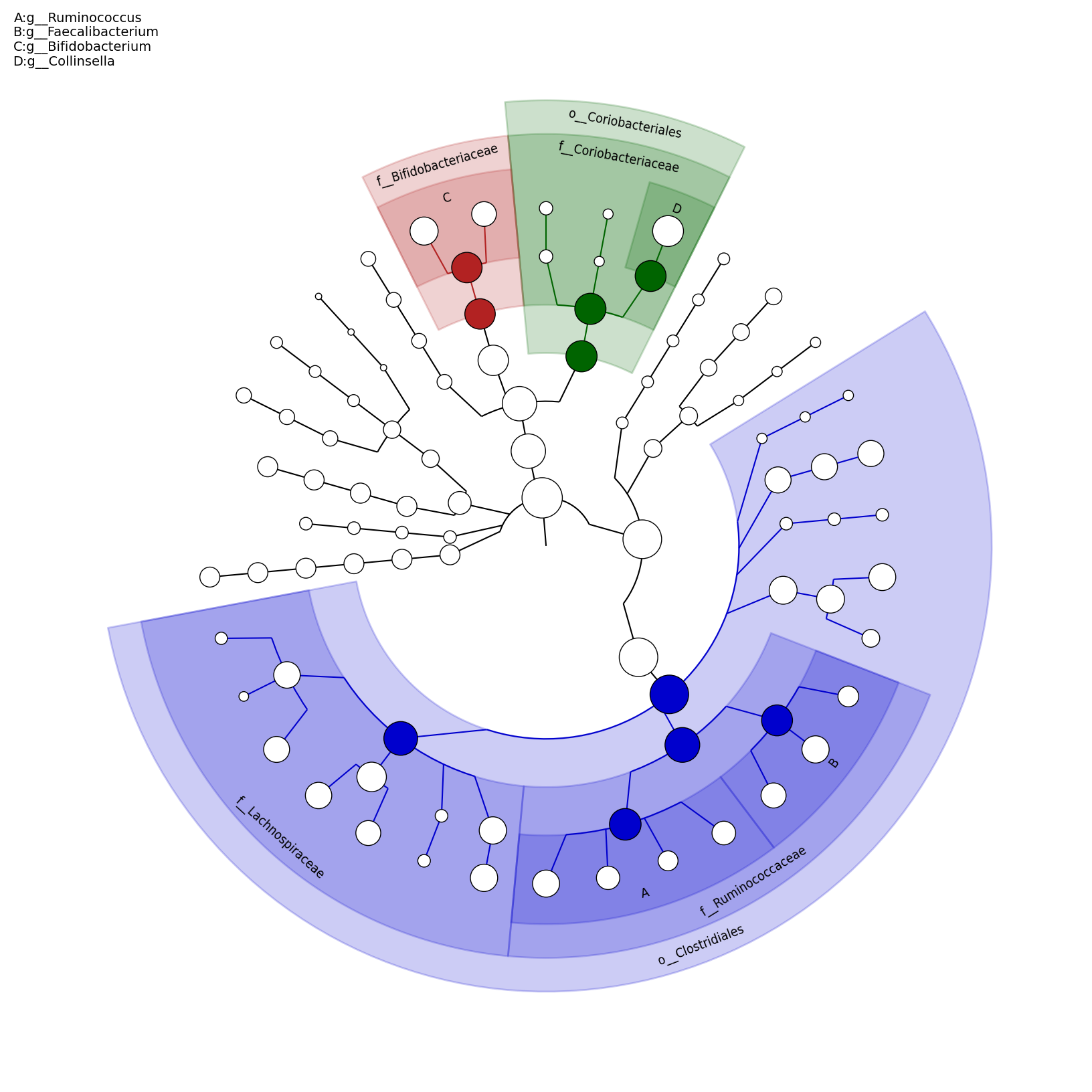
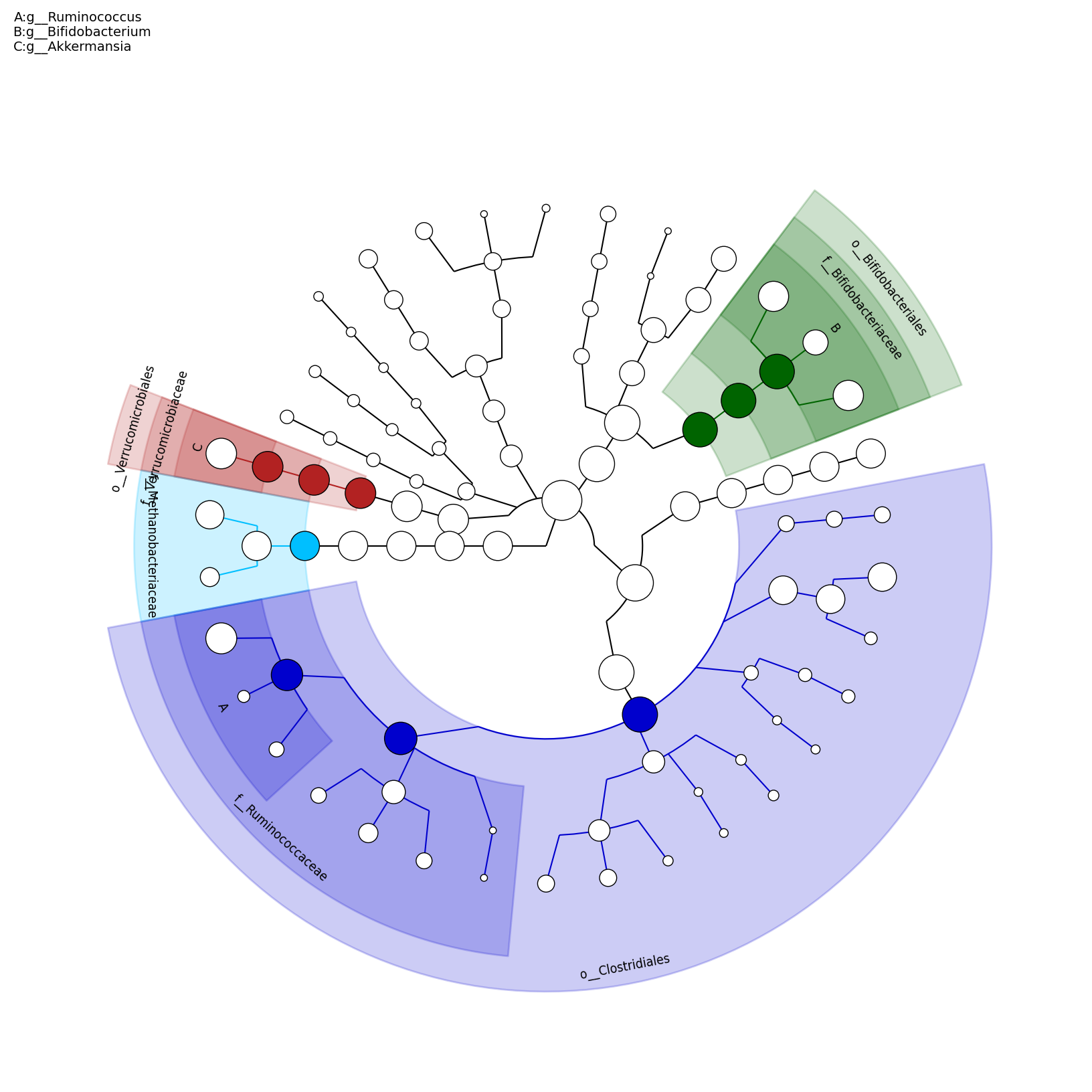
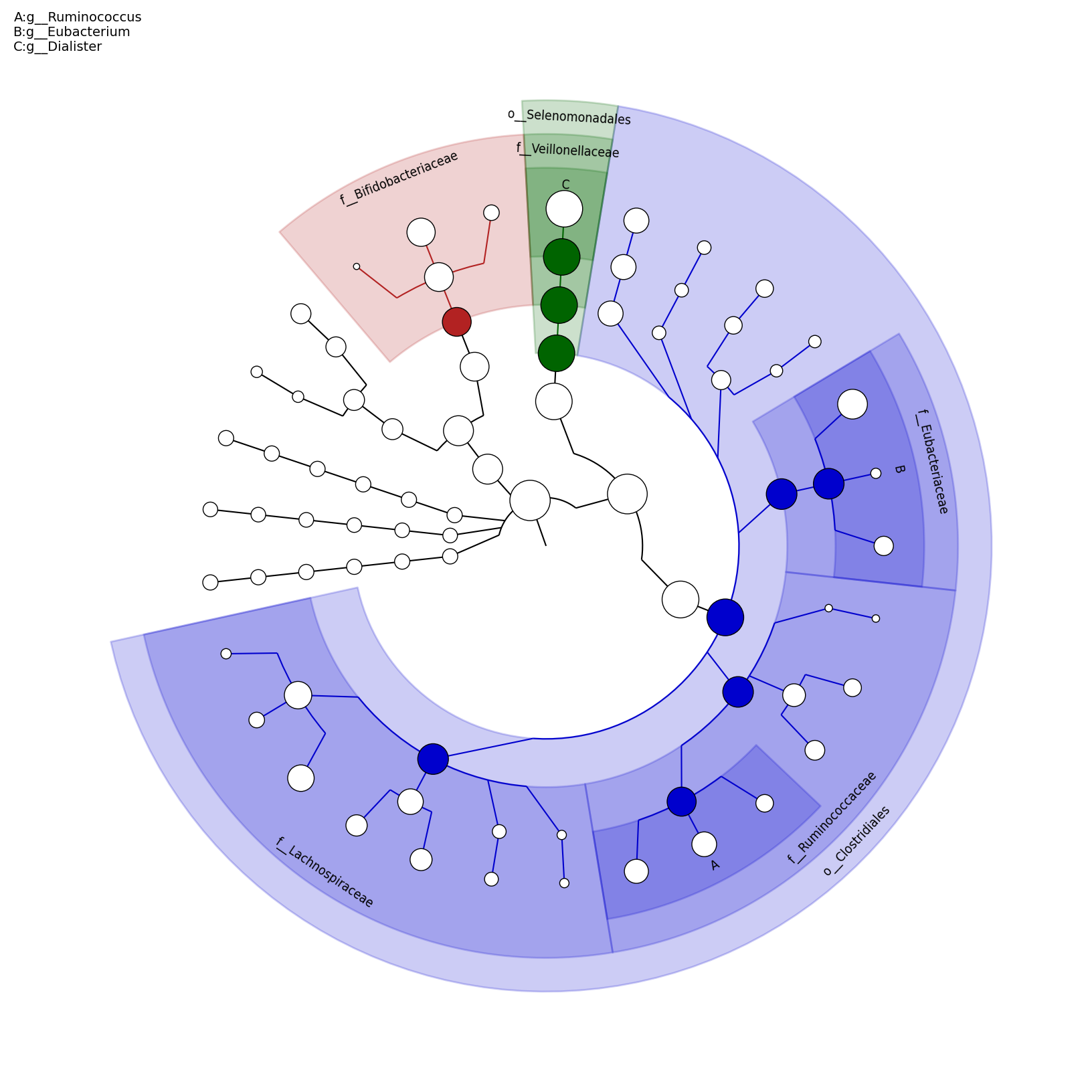
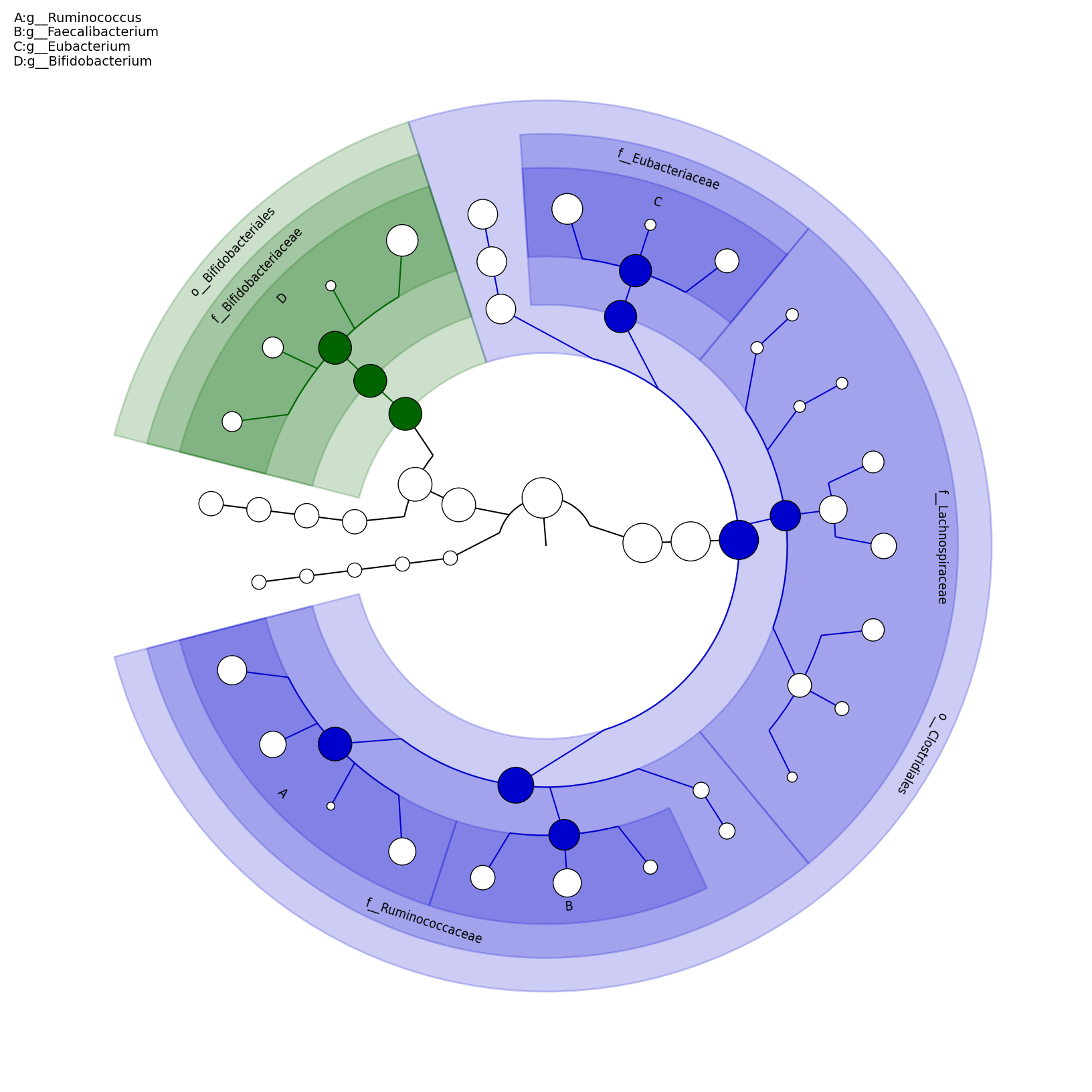
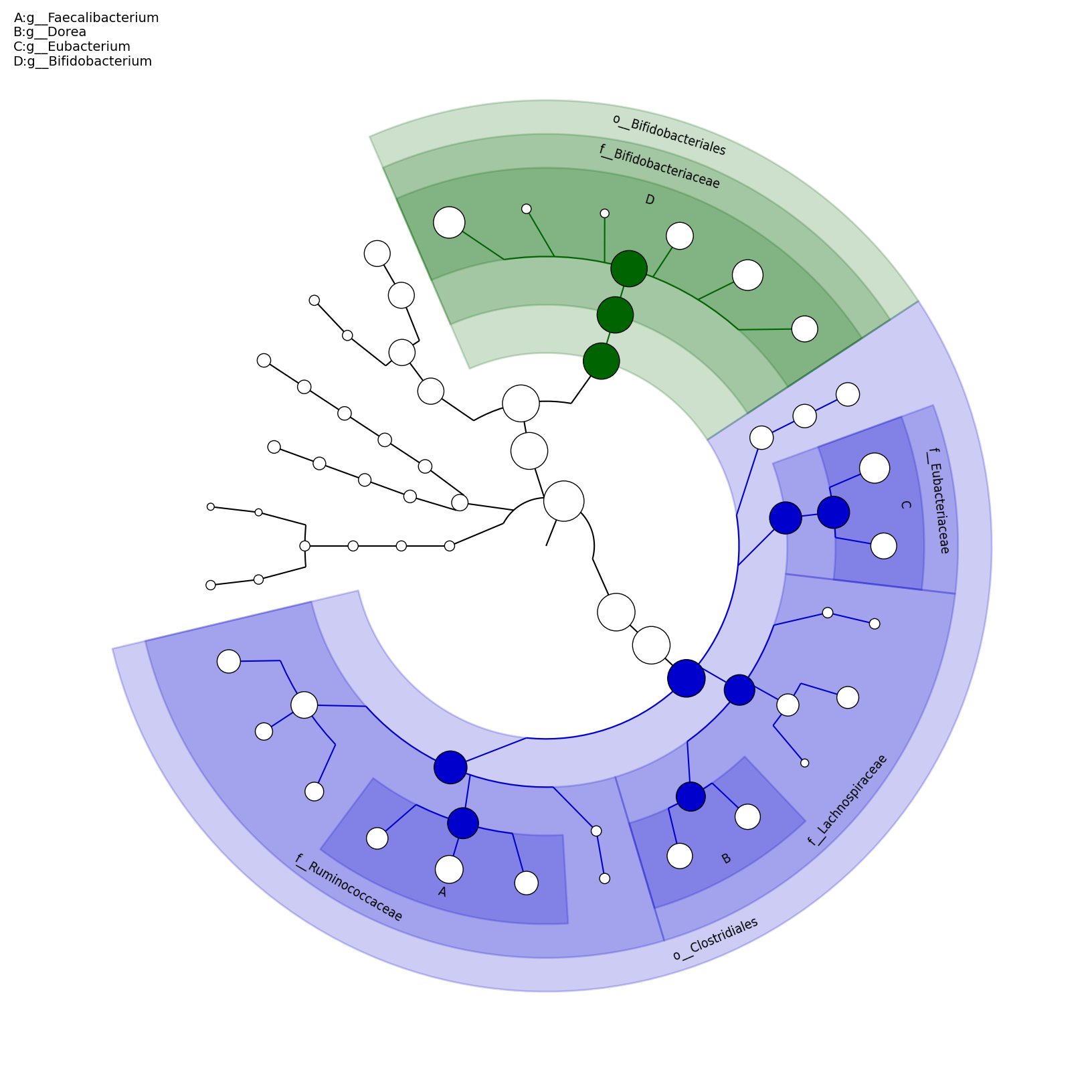
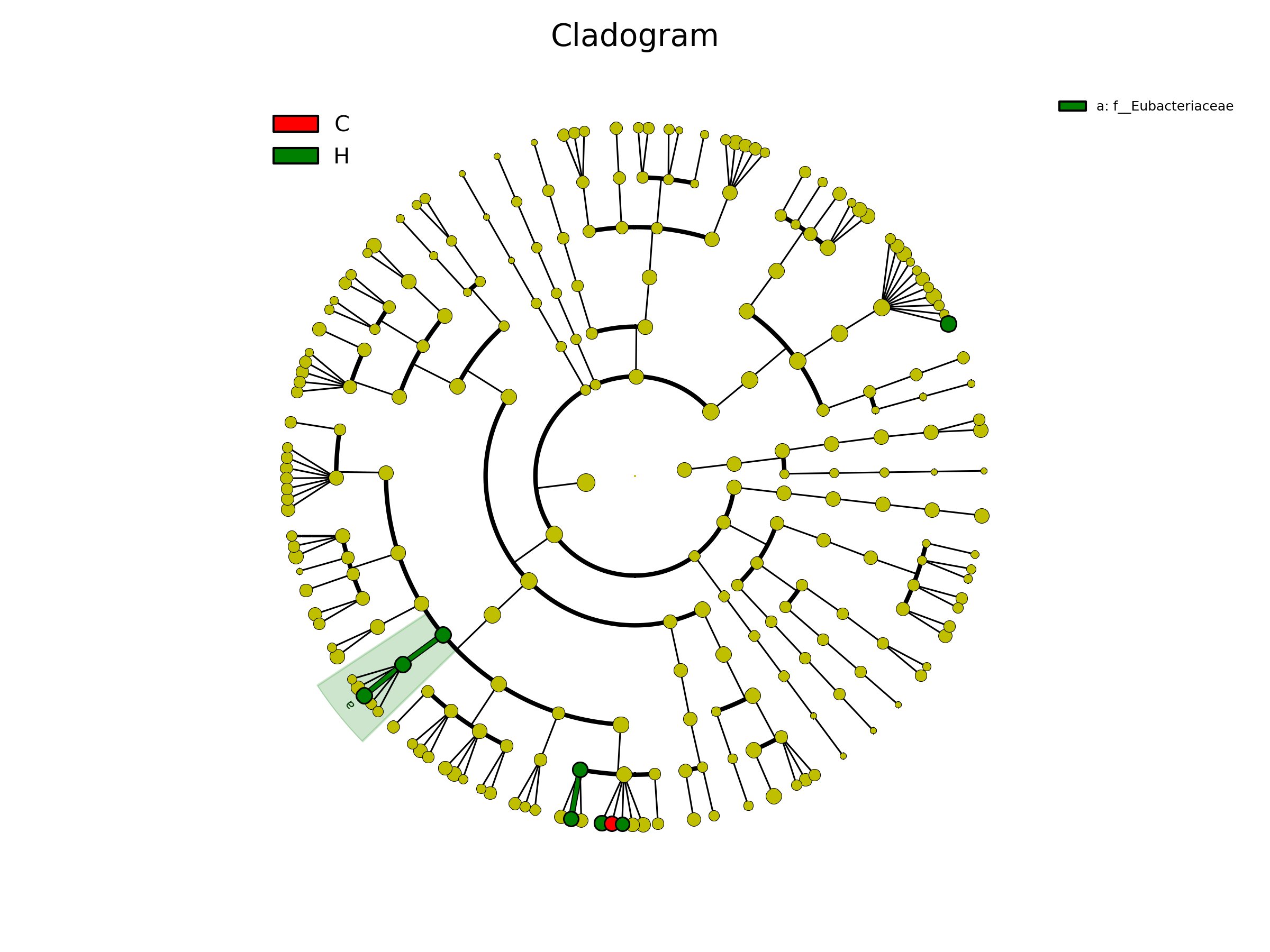
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ python $lefse_dir/plot_cladogram.py --dpi 300 --format png merged_abundance_table.lefse.out lefse_biomarkers_cladogram.png
lefse_biomarkers_cladogram.png
MScBioinf@quince-srv2 ~/MTutorial_Umer]$ python $lefse_dir/plot_features.py -f one --feature_name "k__Bacteria.p__Firmicutes" merged_abundance_table.lefse merged_abundance_table.lefse.out Firmicutes.png [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ python $lefse_dir/plot_features.py -f one --feature_name "k__Bacteria.p__Actinobacteria.c__Actinobacteria.o__Bifidobacteriales.f__Bifidobacteriaceae.g__Bifidobacterium.s__Bifidobacterium_bifidum" merged_abundance_table.lefse merged_abundance_table.lefse.out Bifidobacterium_bifidum.pngFirmicutes.png
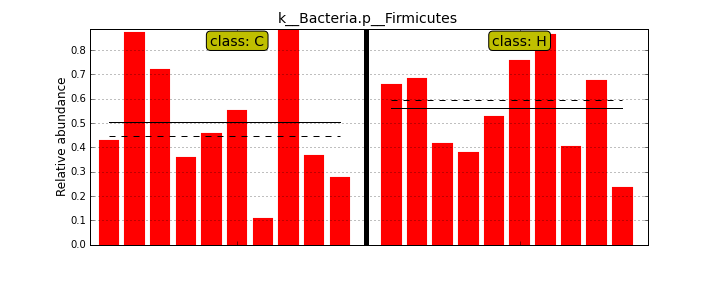
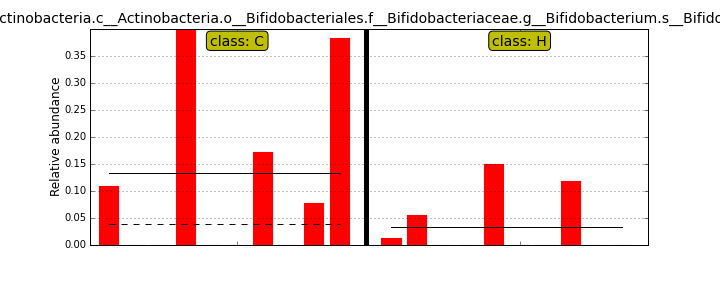
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ cd subsampled_s100000 [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ for i in $(ls * -d); do cd $i; rapsearch -d /home/opt/keggs_database/kegg_db -z 10 -q Raw/${i}_R12.fasta -o ${i}_R12.fasta.out; cd ..; done [MScBioinf@quince-srv2 ~/MTutorial_Umer/subsampled_s100000]$ cd .. [MScBioinf@quince-srv2 ~/MTutorial_Umer]$
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ cp -r /home/opt/Mtutorial/MTutorial_Umer/subsampled_s100000 .
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ cp -r /home/opt/humann . [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ cd humann/input [MScBioinf@quince-srv2 ~/MTutorial_Umer/humann/input]$ cp ../../subsampled_s100000/*/*.fasta.out.m8 . [MScBioinf@quince-srv2 ~/MTutorial_Umer/humann/input]$ ls C13_R12.fasta.out.m8 C27_R12.fasta.out.m8 C45_R12.fasta.out.m8 C56_R12.fasta.out.m8 C68_R12.fasta.out.m8 H117_R12.fasta.out.m8 H128_R12.fasta.out.m8 H132_R12.fasta.out.m8 H139_R12.fasta.out.m8 H142_R12.fasta.out.m8 C1_R12.fasta.out.m8 C32_R12.fasta.out.m8 C51_R12.fasta.out.m8 C62_R12.fasta.out.m8 C80_R12.fasta.out.m8 H119_R12.fasta.out.m8 H130_R12.fasta.out.m8 H134_R12.fasta.out.m8 H140_R12.fasta.out.m8 H144_R12.fasta.out.m8 [MScBioinf@quince-srv2 ~/MTutorial_Umer/humann/input]$We then reformat the file names for HUMAnN to pick them up automatically
[MScBioinf@quince-srv2 ~/MTutorial_Umer/humann/input]$ for i in $(ls *.m8); do mv $i $(echo $i| cut -d"_" -f1).txt; done [MScBioinf@quince-srv2 ~/MTutorial_Umer/humann/input]$ ls C13.txt C1.txt C27.txt C32.txt C45.txt C51.txt C56.txt C62.txt C68.txt C80.txt H117.txt H119.txt H128.txt H130.txt H132.txt H134.txt H139.txt H140.txt H142.txt H144.txt [MScBioinf@quince-srv2 ~/MTutorial_Umer/humann/input]$Now come out of the input folder and run scons with -j 10 switch (i.e. run the analysis on 10 cores)
[MScBioinf@quince-srv2 ~/MTutorial_Umer/humann/input]$ cd .. [MScBioinf@quince-srv2 ~/MTutorial_Umer/humann]$ scons -j 10 scons: Reading SConscript files ... scons: done reading SConscript files. scons: Building targets ... rn(["output/C1_00-hit.txt.gz"], ["input/C1.txt", "src/blast2hits.py"]) /home/MScBioinf/MTutorial_Umer/humann/input/C1.txt cat /home/MScBioinf/MTutorial_Umer/humann/input/C1.txt | src/blast2hits.py | gzip -c | src/output.py output/C1_00-hit.txt.gz rn(["data/koc"], ["data/koc.gz"]) /home/MScBioinf/MTutorial_Umer/humann/data/koc.gz gunzip -c /home/MScBioinf/MTutorial_Umer/humann/data/koc.gz | cat | src/output.py data/koc rn(["data/genels"], ["data/genels.gz"]) /home/MScBioinf/MTutorial_Umer/humann/data/genels.gz gunzip -c /home/MScBioinf/MTutorial_Umer/humann/data/genels.gz | cat | src/output.py data/genels rn(["output/C13_00-hit.txt.gz"], ["input/C13.txt", "src/blast2hits.py"]) /home/MScBioinf/MTutorial_Umer/humann/input/C13.txt cat /home/MScBioinf/MTutorial_Umer/humann/input/C13.txt | src/blast2hits.py | gzip -c | src/output.py output/C13_00-hit.txt.gz rn(["output/C27_00-hit.txt.gz"], ["input/C27.txt", "src/blast2hits.py"]) /home/MScBioinf/MTutorial_Umer/humann/input/C27.txt cat /home/MScBioinf/MTutorial_Umer/humann/input/C27.txt | src/blast2hits.py | gzip -c | src/output.py output/C27_00-hit.txt.gz rn(["output/C32_00-hit.txt.gz"], ["input/C32.txt", "src/blast2hits.py"]) /home/MScBioinf/MTutorial_Umer/humann/input/C32.txt cat /home/MScBioinf/MTutorial_Umer/humann/input/C32.txt | src/blast2hits.py | gzip -c | src/output.py output/C32_00-hit.txt.gz ...When finished, the following files will be generated in the output folder:
[MScBioinf@quince-srv2 ~/MTutorial_Umer/humann]$ ls output C1_00-hit.txt.gz C45_00-hit.txt.gz C68_00-hit.txt.gz H128_00-hit.txt.gz H139_00-hit.txt.gz C1_01b-hit-keg-cat.txt C45_01b-hit-keg-cat.txt C68_01b-hit-keg-cat.txt H128_01b-hit-keg-cat.txt H139_01b-hit-keg-cat.txt C1_01-hit-keg.txt C45_01-hit-keg.txt C68_01-hit-keg.txt H128_01-hit-keg.txt H139_01-hit-keg.txt C1_02a-hit-keg-mpm.txt C45_02a-hit-keg-mpm.txt C68_02a-hit-keg-mpm.txt H128_02a-hit-keg-mpm.txt H139_02a-hit-keg-mpm.txt C1_02a-hit-keg-mpt.txt C45_02a-hit-keg-mpt.txt C68_02a-hit-keg-mpt.txt H128_02a-hit-keg-mpt.txt H139_02a-hit-keg-mpt.txt C1_02b-hit-keg-mpm-cop.txt C45_02b-hit-keg-mpm-cop.txt C68_02b-hit-keg-mpm-cop.txt H128_02b-hit-keg-mpm-cop.txt H139_02b-hit-keg-mpm-cop.txt C1_02b-hit-keg-mpt-cop.txt C45_02b-hit-keg-mpt-cop.txt C68_02b-hit-keg-mpt-cop.txt H128_02b-hit-keg-mpt-cop.txt H139_02b-hit-keg-mpt-cop.txt C1_03a-hit-keg-mpm-cop-nul.txt C45_03a-hit-keg-mpm-cop-nul.txt C68_03a-hit-keg-mpm-cop-nul.txt H128_03a-hit-keg-mpm-cop-nul.txt H139_03a-hit-keg-mpm-cop-nul.txt C1_03a-hit-keg-mpt-cop-nul.txt C45_03a-hit-keg-mpt-cop-nul.txt C68_03a-hit-keg-mpt-cop-nul.txt H128_03a-hit-keg-mpt-cop-nul.txt H139_03a-hit-keg-mpt-cop-nul.txt C1_03b-hit-keg-mpm-cop-nul-nve.txt C45_03b-hit-keg-mpm-cop-nul-nve.txt C68_03b-hit-keg-mpm-cop-nul-nve.txt H128_03b-hit-keg-mpm-cop-nul-nve.txt H139_03b-hit-keg-mpm-cop-nul-nve.txt C1_03b-hit-keg-mpt-cop-nul-nve.txt C45_03b-hit-keg-mpt-cop-nul-nve.txt C68_03b-hit-keg-mpt-cop-nul-nve.txt H128_03b-hit-keg-mpt-cop-nul-nve.txt H139_03b-hit-keg-mpt-cop-nul-nve.txt C1_03c-hit-keg-mpm-cop-nul-nve-nve.txt C45_03c-hit-keg-mpm-cop-nul-nve-nve.txt C68_03c-hit-keg-mpm-cop-nul-nve-nve.txt H128_03c-hit-keg-mpm-cop-nul-nve-nve.txt H139_03c-hit-keg-mpm-cop-nul-nve-nve.txt C1_03c-hit-keg-mpt-cop-nul-nve-nve.txt C45_03c-hit-keg-mpt-cop-nul-nve-nve.txt C68_03c-hit-keg-mpt-cop-nul-nve-nve.txt H128_03c-hit-keg-mpt-cop-nul-nve-nve.txt H139_03c-hit-keg-mpt-cop-nul-nve-nve.txt C1_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt C45_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt C68_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt H128_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt H139_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt C1_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt C45_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt C68_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt H128_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt H139_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt C1_04b-hit-keg-mpm-cop-nul-nve-nve.txt C45_04b-hit-keg-mpm-cop-nul-nve-nve.txt C68_04b-hit-keg-mpm-cop-nul-nve-nve.txt H128_04b-hit-keg-mpm-cop-nul-nve-nve.txt H139_04b-hit-keg-mpm-cop-nul-nve-nve.txt C1_04b-hit-keg-mpt-cop-nul-nve-nve.txt C45_04b-hit-keg-mpt-cop-nul-nve-nve.txt C68_04b-hit-keg-mpt-cop-nul-nve-nve.txt H128_04b-hit-keg-mpt-cop-nul-nve-nve.txt H139_04b-hit-keg-mpt-cop-nul-nve-nve.txt C13_00-hit.txt.gz C51_00-hit.txt.gz C80_00-hit.txt.gz H130_00-hit.txt.gz H140_00-hit.txt.gz C13_01b-hit-keg-cat.txt C51_01b-hit-keg-cat.txt C80_01b-hit-keg-cat.txt H130_01b-hit-keg-cat.txt H140_01b-hit-keg-cat.txt C13_01-hit-keg.txt C51_01-hit-keg.txt C80_01-hit-keg.txt H130_01-hit-keg.txt H140_01-hit-keg.txt C13_02a-hit-keg-mpm.txt C51_02a-hit-keg-mpm.txt C80_02a-hit-keg-mpm.txt H130_02a-hit-keg-mpm.txt H140_02a-hit-keg-mpm.txt C13_02a-hit-keg-mpt.txt C51_02a-hit-keg-mpt.txt C80_02a-hit-keg-mpt.txt H130_02a-hit-keg-mpt.txt H140_02a-hit-keg-mpt.txt C13_02b-hit-keg-mpm-cop.txt C51_02b-hit-keg-mpm-cop.txt C80_02b-hit-keg-mpm-cop.txt H130_02b-hit-keg-mpm-cop.txt H140_02b-hit-keg-mpm-cop.txt C13_02b-hit-keg-mpt-cop.txt C51_02b-hit-keg-mpt-cop.txt C80_02b-hit-keg-mpt-cop.txt H130_02b-hit-keg-mpt-cop.txt H140_02b-hit-keg-mpt-cop.txt C13_03a-hit-keg-mpm-cop-nul.txt C51_03a-hit-keg-mpm-cop-nul.txt C80_03a-hit-keg-mpm-cop-nul.txt H130_03a-hit-keg-mpm-cop-nul.txt H140_03a-hit-keg-mpm-cop-nul.txt C13_03a-hit-keg-mpt-cop-nul.txt C51_03a-hit-keg-mpt-cop-nul.txt C80_03a-hit-keg-mpt-cop-nul.txt H130_03a-hit-keg-mpt-cop-nul.txt H140_03a-hit-keg-mpt-cop-nul.txt C13_03b-hit-keg-mpm-cop-nul-nve.txt C51_03b-hit-keg-mpm-cop-nul-nve.txt C80_03b-hit-keg-mpm-cop-nul-nve.txt H130_03b-hit-keg-mpm-cop-nul-nve.txt H140_03b-hit-keg-mpm-cop-nul-nve.txt C13_03b-hit-keg-mpt-cop-nul-nve.txt C51_03b-hit-keg-mpt-cop-nul-nve.txt C80_03b-hit-keg-mpt-cop-nul-nve.txt H130_03b-hit-keg-mpt-cop-nul-nve.txt H140_03b-hit-keg-mpt-cop-nul-nve.txt C13_03c-hit-keg-mpm-cop-nul-nve-nve.txt C51_03c-hit-keg-mpm-cop-nul-nve-nve.txt C80_03c-hit-keg-mpm-cop-nul-nve-nve.txt H130_03c-hit-keg-mpm-cop-nul-nve-nve.txt H140_03c-hit-keg-mpm-cop-nul-nve-nve.txt C13_03c-hit-keg-mpt-cop-nul-nve-nve.txt C51_03c-hit-keg-mpt-cop-nul-nve-nve.txt C80_03c-hit-keg-mpt-cop-nul-nve-nve.txt H130_03c-hit-keg-mpt-cop-nul-nve-nve.txt H140_03c-hit-keg-mpt-cop-nul-nve-nve.txt C13_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt C51_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt C80_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt H130_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt H140_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt C13_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt C51_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt C80_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt H130_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt H140_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt C13_04b-hit-keg-mpm-cop-nul-nve-nve.txt C51_04b-hit-keg-mpm-cop-nul-nve-nve.txt C80_04b-hit-keg-mpm-cop-nul-nve-nve.txt H130_04b-hit-keg-mpm-cop-nul-nve-nve.txt H140_04b-hit-keg-mpm-cop-nul-nve-nve.txt C13_04b-hit-keg-mpt-cop-nul-nve-nve.txt C51_04b-hit-keg-mpt-cop-nul-nve-nve.txt C80_04b-hit-keg-mpt-cop-nul-nve-nve.txt H130_04b-hit-keg-mpt-cop-nul-nve-nve.txt H140_04b-hit-keg-mpt-cop-nul-nve-nve.txt C27_00-hit.txt.gz C56_00-hit.txt.gz H117_00-hit.txt.gz H132_00-hit.txt.gz H142_00-hit.txt.gz C27_01b-hit-keg-cat.txt C56_01b-hit-keg-cat.txt H117_01b-hit-keg-cat.txt H132_01b-hit-keg-cat.txt H142_01b-hit-keg-cat.txt C27_01-hit-keg.txt C56_01-hit-keg.txt H117_01-hit-keg.txt H132_01-hit-keg.txt H142_01-hit-keg.txt C27_02a-hit-keg-mpm.txt C56_02a-hit-keg-mpm.txt H117_02a-hit-keg-mpm.txt H132_02a-hit-keg-mpm.txt H142_02a-hit-keg-mpm.txt C27_02a-hit-keg-mpt.txt C56_02a-hit-keg-mpt.txt H117_02a-hit-keg-mpt.txt H132_02a-hit-keg-mpt.txt H142_02a-hit-keg-mpt.txt C27_02b-hit-keg-mpm-cop.txt C56_02b-hit-keg-mpm-cop.txt H117_02b-hit-keg-mpm-cop.txt H132_02b-hit-keg-mpm-cop.txt H142_02b-hit-keg-mpm-cop.txt C27_02b-hit-keg-mpt-cop.txt C56_02b-hit-keg-mpt-cop.txt H117_02b-hit-keg-mpt-cop.txt H132_02b-hit-keg-mpt-cop.txt H142_02b-hit-keg-mpt-cop.txt C27_03a-hit-keg-mpm-cop-nul.txt C56_03a-hit-keg-mpm-cop-nul.txt H117_03a-hit-keg-mpm-cop-nul.txt H132_03a-hit-keg-mpm-cop-nul.txt H142_03a-hit-keg-mpm-cop-nul.txt C27_03a-hit-keg-mpt-cop-nul.txt C56_03a-hit-keg-mpt-cop-nul.txt H117_03a-hit-keg-mpt-cop-nul.txt H132_03a-hit-keg-mpt-cop-nul.txt H142_03a-hit-keg-mpt-cop-nul.txt C27_03b-hit-keg-mpm-cop-nul-nve.txt C56_03b-hit-keg-mpm-cop-nul-nve.txt H117_03b-hit-keg-mpm-cop-nul-nve.txt H132_03b-hit-keg-mpm-cop-nul-nve.txt H142_03b-hit-keg-mpm-cop-nul-nve.txt C27_03b-hit-keg-mpt-cop-nul-nve.txt C56_03b-hit-keg-mpt-cop-nul-nve.txt H117_03b-hit-keg-mpt-cop-nul-nve.txt H132_03b-hit-keg-mpt-cop-nul-nve.txt H142_03b-hit-keg-mpt-cop-nul-nve.txt C27_03c-hit-keg-mpm-cop-nul-nve-nve.txt C56_03c-hit-keg-mpm-cop-nul-nve-nve.txt H117_03c-hit-keg-mpm-cop-nul-nve-nve.txt H132_03c-hit-keg-mpm-cop-nul-nve-nve.txt H142_03c-hit-keg-mpm-cop-nul-nve-nve.txt C27_03c-hit-keg-mpt-cop-nul-nve-nve.txt C56_03c-hit-keg-mpt-cop-nul-nve-nve.txt H117_03c-hit-keg-mpt-cop-nul-nve-nve.txt H132_03c-hit-keg-mpt-cop-nul-nve-nve.txt H142_03c-hit-keg-mpt-cop-nul-nve-nve.txt C27_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt C56_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt H117_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt H132_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt H142_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt C27_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt C56_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt H117_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt H132_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt H142_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt C27_04b-hit-keg-mpm-cop-nul-nve-nve.txt C56_04b-hit-keg-mpm-cop-nul-nve-nve.txt H117_04b-hit-keg-mpm-cop-nul-nve-nve.txt H132_04b-hit-keg-mpm-cop-nul-nve-nve.txt H142_04b-hit-keg-mpm-cop-nul-nve-nve.txt C27_04b-hit-keg-mpt-cop-nul-nve-nve.txt C56_04b-hit-keg-mpt-cop-nul-nve-nve.txt H117_04b-hit-keg-mpt-cop-nul-nve-nve.txt H132_04b-hit-keg-mpt-cop-nul-nve-nve.txt H142_04b-hit-keg-mpt-cop-nul-nve-nve.txt C32_00-hit.txt.gz C62_00-hit.txt.gz H119_00-hit.txt.gz H134_00-hit.txt.gz H144_00-hit.txt.gz C32_01b-hit-keg-cat.txt C62_01b-hit-keg-cat.txt H119_01b-hit-keg-cat.txt H134_01b-hit-keg-cat.txt H144_01b-hit-keg-cat.txt C32_01-hit-keg.txt C62_01-hit-keg.txt H119_01-hit-keg.txt H134_01-hit-keg.txt H144_01-hit-keg.txt C32_02a-hit-keg-mpm.txt C62_02a-hit-keg-mpm.txt H119_02a-hit-keg-mpm.txt H134_02a-hit-keg-mpm.txt H144_02a-hit-keg-mpm.txt C32_02a-hit-keg-mpt.txt C62_02a-hit-keg-mpt.txt H119_02a-hit-keg-mpt.txt H134_02a-hit-keg-mpt.txt H144_02a-hit-keg-mpt.txt C32_02b-hit-keg-mpm-cop.txt C62_02b-hit-keg-mpm-cop.txt H119_02b-hit-keg-mpm-cop.txt H134_02b-hit-keg-mpm-cop.txt H144_02b-hit-keg-mpm-cop.txt C32_02b-hit-keg-mpt-cop.txt C62_02b-hit-keg-mpt-cop.txt H119_02b-hit-keg-mpt-cop.txt H134_02b-hit-keg-mpt-cop.txt H144_02b-hit-keg-mpt-cop.txt C32_03a-hit-keg-mpm-cop-nul.txt C62_03a-hit-keg-mpm-cop-nul.txt H119_03a-hit-keg-mpm-cop-nul.txt H134_03a-hit-keg-mpm-cop-nul.txt H144_03a-hit-keg-mpm-cop-nul.txt C32_03a-hit-keg-mpt-cop-nul.txt C62_03a-hit-keg-mpt-cop-nul.txt H119_03a-hit-keg-mpt-cop-nul.txt H134_03a-hit-keg-mpt-cop-nul.txt H144_03a-hit-keg-mpt-cop-nul.txt C32_03b-hit-keg-mpm-cop-nul-nve.txt C62_03b-hit-keg-mpm-cop-nul-nve.txt H119_03b-hit-keg-mpm-cop-nul-nve.txt H134_03b-hit-keg-mpm-cop-nul-nve.txt H144_03b-hit-keg-mpm-cop-nul-nve.txt C32_03b-hit-keg-mpt-cop-nul-nve.txt C62_03b-hit-keg-mpt-cop-nul-nve.txt H119_03b-hit-keg-mpt-cop-nul-nve.txt H134_03b-hit-keg-mpt-cop-nul-nve.txt H144_03b-hit-keg-mpt-cop-nul-nve.txt C32_03c-hit-keg-mpm-cop-nul-nve-nve.txt C62_03c-hit-keg-mpm-cop-nul-nve-nve.txt H119_03c-hit-keg-mpm-cop-nul-nve-nve.txt H134_03c-hit-keg-mpm-cop-nul-nve-nve.txt H144_03c-hit-keg-mpm-cop-nul-nve-nve.txt C32_03c-hit-keg-mpt-cop-nul-nve-nve.txt C62_03c-hit-keg-mpt-cop-nul-nve-nve.txt H119_03c-hit-keg-mpt-cop-nul-nve-nve.txt H134_03c-hit-keg-mpt-cop-nul-nve-nve.txt H144_03c-hit-keg-mpt-cop-nul-nve-nve.txt C32_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt C62_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt H119_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt H134_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt H144_04a-hit-keg-mpm-cop-nul-nve-nve-xpe.txt C32_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt C62_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt H119_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt H134_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt H144_04a-hit-keg-mpt-cop-nul-nve-nve-xpe.txt C32_04b-hit-keg-mpm-cop-nul-nve-nve.txt C62_04b-hit-keg-mpm-cop-nul-nve-nve.txt H119_04b-hit-keg-mpm-cop-nul-nve-nve.txt H134_04b-hit-keg-mpm-cop-nul-nve-nve.txt H144_04b-hit-keg-mpm-cop-nul-nve-nve.txt C32_04b-hit-keg-mpt-cop-nul-nve-nve.txt C62_04b-hit-keg-mpt-cop-nul-nve-nve.txt H119_04b-hit-keg-mpt-cop-nul-nve-nve.txt H134_04b-hit-keg-mpt-cop-nul-nve-nve.txt H144_04b-hit-keg-mpt-cop-nul-nve-nve.txt
| *_00-*.txt.gz A condensed binary representation of translated BLAST results, abstracted from and independent of the specific format (blastx/mblastx/mapx) in which they are provided. |
|
*_01-*.txt Generated from *_00-*.txt.gz Relative gene abundances as calculated from BLAST results in which each read has been mapped to zero or more gene identifiers based on quality of match. Total weight of each read is 1.0, distributed over all gene (KO) matches by quality. |
|
*_02a-*.txt Generated from *_01-*.txt Relative gene abundances distributed over all pathways in which the gene is predicted to occur. |
|
*_02b-*.txt Generated from *_02a-*.txt Relative gene abundances with pathway assignments, taxonomically limited to remove pathways that could only occur in low-abundance/absent organisms. |
|
*_03a-*.txt Generated from *_02b-*.txt Relative gene abundances with pathway assignments, smoothed so that zero means zero and non-zero values are imputed to account for non-observed sequences. |
|
*_03b-*.txt Generated from *_03a-*.txt Relative gene abundances with pathway assignments, gap-filled so that gene/pathway combinations with surprisingly low frequency are imputed to contain a more plausible value. |
|
*_04a-*.txt Generated from *_03b-*.txt Pathway coverage (presence/absence) measure, i.e. relative confidence of each pathway being present in the sample. Values are between 0 and 1 inclusive. |
|
*_04b-*.txt Generated from *_03b-*.txt Pathway abundance measure, i.e. relative "copy number" of each pathway in the sample. On the same relative abundance scale (0 and up) as the original gene abundances _01-*.txt. |
|
*_99-*.txt Generated from *_00-*.txt.gz Per-sample gene abundance tables formatted for loading into METAREP. On the same relative abundance scale (0 and up) as the original gene abundances _01-*.txt. |
| 04a-*.txt Combined pathway coverage matrix for all samples. |
|
04b-*.txt Combined pathway abundance matrix for all samples, normalized per column. |
|
04b-*-lefse.txt Pathway information formatted for input to LefSe (exported) |
|
04b-*-graphlan_tree.txt Pathway information formatted as a tree reflecting the KEGG BRITE hierarchy for plotting in GraPhlAn |
|
04b-*-graphlan_rings.txt Pathway abundances formatted for overplotting on the KEGG BRITE hierarchy using GraPhlAn |
|
01b-*.txt Combined KO abundance matrix for all samples, normalized per column |
|
*_01-keg*.txt Gene abundance calculated as the confidence (e-value/p-value) weighted sum of all hits for each read. |
|
*_02*-mpt*.txt Gene to pathway assignment performed using MinPath. |
|
*_02*-nve*.txt Gene to pathway assignment performed naively using all pathways. |
|
*_02*-cop*.txt Pathway abundances adjusted based on A) taxonomic limitation and B) the expected copy number of each gene in the detected organisms. |
|
*_03a*-wbl*.txt Smoothing performed using Witten-Bell discounting, which shifts sum_observed/(sum_observed + num_observed) probability mass into zero counts and reduces others by the same fraction. |
|
*_03a*-nve*.txt Smoothing performed naively by adding a constant value (0.1) to missing gene/pathway combinations. |
|
*_03b*-nul*.txt No gap filling (no-operation, abundances left as is). |
|
*_03b*-nve*.txt Gap filling by substituting any values below each pathway's median with the median value itself. |
|
*_04a*-nve*.txt Pathway coverage calculated as fraction of genes in pathway at or above global median abundance. |
|
*_04a*-xpe*.txt Pathway coverage calculated as fraction of genes in pathway at or above global median abundance, with low-abundance pathways set to zero using Xipe. |
|
*_04b*-nve*.txt Pathway abundance calculated as average abundance of the most abundant half of genes in the pathway. |
[MScBioinf@quince-srv2 ~/MTutorial_Umer/humann]$ cd .. [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ $metaphlan_dir/utils/merge_metaphlan_tables.py humann/output/*_04b-hit-keg-mpm-cop-nul-nve-nve.txt | sed -e 's/\([CH]\)[0-9]*_04b-hit-keg-mpm-cop-nul-nve-nve/\1/g' | awk '!/^PID/{print}' > merged_humann_abundance_table.lefse.IDs.txt [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ head merged_humann_abundance_table.lefse.IDs.txt ID C C C C C C C C C C H H H H H H H H H H M00001 63.3419969282 71.607578932 66.1026404876 64.3500136419 69.3295201772 70.3131234989 73.7764512813 69.0464301663 77.2585689034 67.8037315438 78.1296335495 68.1654721135 74.2196040709 66.3114414775 60.662945951 75.5256245735 81.5951407048 65.7851448774 65.9269560207 75.670172475 M00002 56.7897204794 63.2949129517 57.8764830639 60.2369255474 64.0703649133 68.0965671655 64.486358436 63.645451366 80.9027854731 61.260906232 76.0086231152 61.0160043476 73.9171206965 64.5866301044 55.3284979236 69.9685422403 74.7490610367 65.5096036261 63.0982773074 73.4602215243 M00003 55.293266528 60.3835293477 56.3792480461 41.8107289044 59.1247224421 49.9797648607 53.6737093134 55.7638335713 44.8498637095 53.8151215307 57.7403520156 56.5110012496 65.0884332643 54.0596586456 55.0854082603 64.1775154856 63.6372857005 52.9701678233 50.8417697141 68.3143774357 M00004 22.7138709984 36.8856390209 48.7304810535 27.4413132949 9.82872002809 10.6268783932 35.3335120958 29.1197850824 43.9435836662 30.4300858622 49.0963588071 12.6471890664 30.5323983132 16.6313799963 17.4073002575 29.2123455227 49.3288770253 10.8295552862 23.3737704764 23.2076814314 M00005 106.921549595 80.1814759562 75.1148130506 66.8059838667 83.6554902195 71.9149212497 90.4020748308 54.2686116516 94.0820581804 70.8883397461 69.7623750113 71.0411967696 67.6345691936 55.1031702176 78.3081081961 80.9352514938 89.4117269745 84.0466522545 66.436088254 69.5421131927 M00006 14.3047217646 24.6739476558 35.8474551111 19.8850532222 5.16832530731 6.19857562356 23.794901164 19.497541262 55.7423643404 20.4574875772 39.8330997777 7.16732989048 21.4469652686 9.38877580591 10.326870282 19.4899452656 35.9092810221 5.99018888399 14.4711710975 14.2593479081 M00007 54.9542803044 73.0267329763 76.0682230439 44.2581532544 74.9201282036 35.474172887 68.5958918862 57.4733811377 36.2669937678 59.3670391458 63.9735146338 51.4515333735 52.9708992246 71.0827362114 54.6051232443 58.2823500763 78.7634183407 52.0155361833 60.6466103115 62.0964250384 M00008 2.79543727905 2.30024868436 14.8390166423 6.92865146945 1.9346130458 0.984655062514 0.0875950008344 4.14671958844 5.04488571105 8.64756311388 6.20121752833 2.15795765178e-05 0.0317375663836 2.05584112246 2.54625576239 2.23959940629 0.00259351604235 0.00120452715802 0.563473385651 0.160190408461 M00009 0.0109939562021 10.9804548012 11.4518977761 0 8.30910073636 9.06083419625 6.21343883676e-12 22.7916691412 10.4381408212 3.3408632852e-07 0.11567990577 0.111679327965 18.4376864926 7.81328796398 0.639518209773 5.26716398396 0.657871923717 0.167872609207 1.63157814012 9.73339500033 [MScBioinf@quince-srv2 ~/MTutorial_Umer]$We will replace IDs given in the first column with the titles. To do this, we will use my perl script, replaceIDs.pl that takes as input the IDs to title mapping.
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ perl /home/opt/Mtutorial/bin/replaceIDs.pl -i merged_humann_abundance_table.lefse.IDs.txt --in_type tab -l humann/data/map_kegg.txt --list_type tab -c 1 > merged_humann_abundance_table.lefse.txt [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ head merged_humann_abundance_table.lefse.txt ID C C C C C C C C C C H H H H H H H H H H Glycolysis (Embden-Meyerhof pathway), glucose => pyruvate 63.3419969282 71.607578932 66.1026404876 64.3500136419 69.3295201772 70.3131234989 73.7764512813 69.0464301663 77.2585689034 67.8037315438 78.1296335495 68.1654721135 74.2196040709 66.3114414775 60.662945951 75.5256245735 81.5951407048 65.7851448774 65.9269560207 75.670172475 Glycolysis, core module involving three-carbon compounds 56.7897204794 63.2949129517 57.8764830639 60.2369255474 64.0703649133 68.0965671655 64.486358436 63.645451366 80.9027854731 61.260906232 76.0086231152 61.0160043476 73.9171206965 64.5866301044 55.3284979236 69.9685422403 74.7490610367 65.5096036261 63.0982773074 73.4602215243 Gluconeogenesis, oxaloacetate => fructose-6P 55.293266528 60.3835293477 56.3792480461 41.8107289044 59.1247224421 49.9797648607 53.6737093134 55.7638335713 44.8498637095 53.8151215307 57.7403520156 56.5110012496 65.0884332643 54.0596586456 55.0854082603 64.1775154856 63.6372857005 52.9701678233 50.8417697141 68.3143774357 Pentose phosphate pathway (Pentose phosphate cycle) 22.7138709984 36.8856390209 48.7304810535 27.4413132949 9.82872002809 10.6268783932 35.3335120958 29.1197850824 43.9435836662 30.4300858622 49.0963588071 12.6471890664 30.5323983132 16.6313799963 17.4073002575 29.2123455227 49.3288770253 10.8295552862 23.3737704764 23.2076814314 PRPP biosynthesis, ribose 5P => PRPP 106.921549595 80.1814759562 75.1148130506 66.8059838667 83.6554902195 71.9149212497 90.4020748308 54.2686116516 94.0820581804 70.8883397461 69.7623750113 71.0411967696 67.6345691936 55.1031702176 78.3081081961 80.9352514938 89.4117269745 84.0466522545 66.436088254 69.5421131927 Pentose phosphate pathway, oxidative phase, glucose 6P => ribulose 5P 14.3047217646 24.6739476558 35.8474551111 19.8850532222 5.16832530731 6.19857562356 23.794901164 19.497541262 55.7423643404 20.4574875772 39.8330997777 7.16732989048 21.4469652686 9.38877580591 10.326870282 19.4899452656 35.9092810221 5.99018888399 14.4711710975 14.2593479081 Pentose phosphate pathway, non-oxidative phase, fructose 6P => ribose 5P 54.9542803044 73.0267329763 76.0682230439 44.2581532544 74.9201282036 35.474172887 68.5958918862 57.4733811377 36.2669937678 59.3670391458 63.9735146338 51.4515333735 52.9708992246 71.0827362114 54.6051232443 58.2823500763 78.7634183407 52.0155361833 60.6466103115 62.0964250384 Entner-Doudoroff pathway, glucose-6P => glyceraldehyde-3P + pyruvate 2.79543727905 2.30024868436 14.8390166423 6.92865146945 1.9346130458 0.984655062514 0.0875950008344 4.14671958844 5.04488571105 8.64756311388 6.20121752833 2.15795765178e-05 0.0317375663836 2.05584112246 2.54625576239 2.23959940629 0.00259351604235 0.00120452715802 0.563473385651 0.160190408461 Citrate cycle (TCA cycle, Krebs cycle) 0.0109939562021 10.9804548012 11.4518977761 0 8.30910073636 9.06083419625 6.21343883676e-12 22.7916691412 10.4381408212 3.3408632852e-07 0.11567990577 0.111679327965 18.4376864926 7.81328796398 0.639518209773 5.26716398396 0.657871923717 0.167872609207 1.63157814012 9.73339500033 [MScBioinf@quince-srv2 ~/MTutorial_Umer]$Then, we will follow the same steps as we did in the previous section, but using merged_humann_abundance_table.lefse.txt and setting LDA threshold to 3.0. This will generate lefse_kegg.png that will display significant pathways with effect sizes.
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ python $lefse_dir/format_input.py merged_humann_abundance_table.lefse.txt merged_humann_abundance_table.lefse -c 1 -o 1000000 [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ python $lefse_dir/run_lefse.py merged_humann_abundance_table.lefse merged_humann_abundance_table.lefse.out -l 3 Number of significantly discriminative features: 40 ( 42 ) before internal wilcoxon Number of discriminative features with abs LDA score > 3.0 : 17 [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ python $lefse_dir/plot_res.py --dpi 300 merged_humann_abundance_table.lefse.out lefse_kegg.png [MScBioinf@quince-srv2 ~/MTutorial_Umer]$lefse_kegg.png
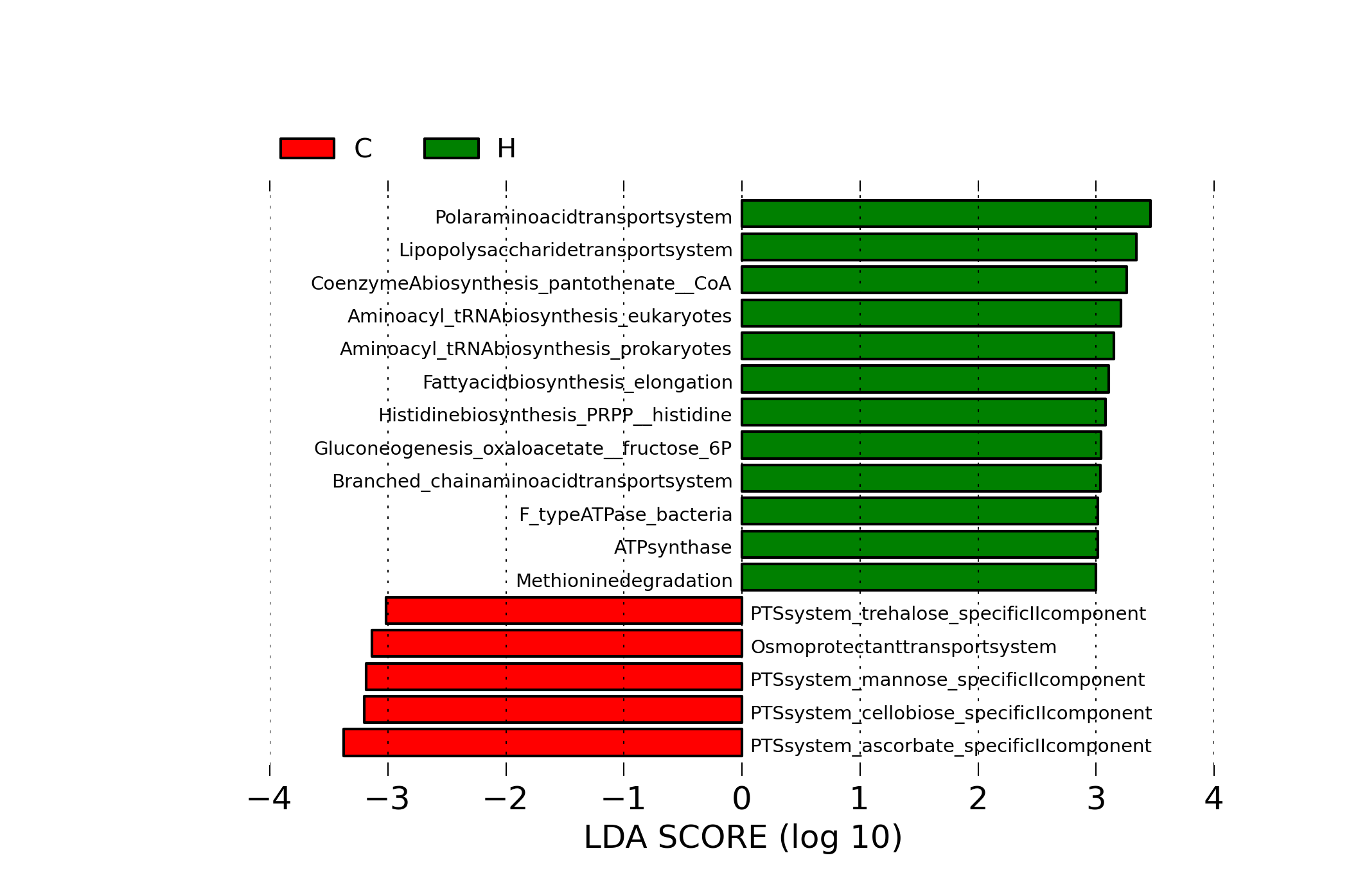
[MScBioinf@quince-srv2 ~/MTutorial_Umer]$ rm -r humann [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ source /home/opt/Mtutorial/bin/disable_python2.7_lefse_rpy2.sh [MScBioinf@quince-srv2 ~/MTutorial_Umer]$ exitIn the analysis done so far, you will notice, that the visualizations are mostly sparse because we considered a small number of reads just to complete this much analysis in time. To get better results, restart the analysis but by creating a new subsampled_s1000000 folder, and subsampling the reads to 1 million! Other than changing the folder name and specifying the subsampling parameter in one command, the flow of instructions will remain same!