$path variable.pcregrep is installed which uses the switch --buffer-size to change the size of internal buffers. For extracting longer contigs, we have to increase the size from default 20K to a higher value. We will set the alias FASTAgrep that will help us in extracting sequences from FASTA file based on a supplied pattern (You can also add this alias to ~/.bashrc):
$ alias FASTAgrep="awk '{gsub(\"_\",\"\\\_\",\$0);\$0=\"(?s)^>\"\$0\".*?(?=\\\n(\\\z|>))\"}1' | pcregrep -oM -f -"
.sh extension and are primarly designed to work with metagenomic contigs.STDIN using less <&0 and unless otherwise specified,take ONLY one argument of space-delimited parameters list in inverted commas that are subsequently passed to the corresponding EMBOSS utility. Accepting sequences from STDIN makes it easier to incorporate FASTAgrep and also helps in integrating GNU's parallel which reduces the execution time. For example, here is the execution time for both sequential and parallel mode for compseq.sh on a given contigs.fa file for contigs > 1000bp:
$ time echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./compseq.sh 2>&1 > /dev/null
real 0m30.686s
user 0m22.476s
sys 0m9.250s
$ time echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | parallel -kN300 --recstart '>' --pipe ./compseq.sh 2>&1 > /dev/null
real 0m6.420s
user 0m25.586s
sys 0m12.203s
Here parallel -kN300 --recstart '>' --pipe splits the input FASTA stream into 300 records (-N) per processor using ">" (--recstart '>') as a delimiter while maintaining the same order (-k) as input stream.
[Contig]\t[Feature]\t[Value], then you can pipe the stream to GENERATEtable.sh:
$ cat test.tsv contig1 F1 12.2 contig1 F2 34.2 contig1 F3 45.2 contig2 F2 56.3 contig2 F3 56.2 contig3 F1 45.4 contig3 F2 56.3 contig4 F1 23.5 contig5 F1 24.5 $ cat GENERATEtable.sh #!/bin/bash less <&0| \ perl -ane '$r{$F[0].":".$F[1]}=$F[2]; unless($F[0]~~@s){ push @s,$F[0];} unless($F[1]~~@m){ push @m,$F[1];} END{ print "Contigs\t".join("\t",@s)."\n"; for($i=0;$i<@m;$i++){ print $m[$i]; for($j=0;$j<@s;$j++){ (not defined $r{$s[$j].":".$m[$i]})?print "\t".0:print"\t".$r{$s[$j].":".$m[$i]};} print "\n";}}' $ cat test.tsv | ./GENERATEtable.sh Contigs contig1 contig2 contig3 contig4 contig5 F1 12.2 0 45.4 23.5 24.5 F2 34.2 56.3 56.3 0 0 F3 45.2 56.2 0 0 0
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' to linearise the sequences. If the input stream is always linearised, then this piece of code can be removed from the shell scripts to speed them up.
-s switch, we can pass arguments to the perl one-liner, for example, the string -- -o=$1 followed by the one-liner passes parameters to the original program in $r=qx/PROGNAME -sequence=asis:$a[1] $o -stdout -auto /;. Since we have written the parsers for a particular output (in most cases standard) format, the wrappers will work only with a subset of parameters given in PROGNAME -help -verbose and are listed next to each wrapper.
compseq.sh: calculate the composition of unique words in sequences
Content of compseq.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/compseq -sequence=asis:$a[1] $o -stdout -auto /;
$r=~s/#.*\n//g;
$r=~s/\s+\n//g;
$r=~s/^Word size\s+\d\nTotal count\s+\d+//g;
$r=~s/Other.*?\n//g;
$r=~s/\n\s+/\n/g;
$r=~s/\h+/\t/g;
if (defined $r and length $r){
print substr($a[0],1)."\t".join("\n".substr($a[0],1)."\t",split("\n",$r))."\n"}}' -- -o=$1
Parameters [from compseq -help -verbose]:
-word integer [2] This is the size of word (n-mer) to
count.
Thus if you want to count codon frequencies
for a nucleotide sequence, you should enter
3 here. (Integer 1 or more)
-frame integer [0] The normal behaviour of 'compseq' is to
count the frequencies of all words that
occur by moving a window of length 'word' up
by one each time.
This option allows you to move the window up
by the length of the word each time,
skipping over the intervening words.
You can count only those words that occur in
a single frame of the word by setting this
value to a number other than zero.
If you set it to 1 it will only count the
words in frame 1, 2 will only count the
words in frame 2 and so on. (Integer 0 or
more)
-[no]ignorebz boolean [Y] The amino acid code B represents
Asparagine or Aspartic acid and the code Z
represents Glutamine or Glutamic acid.
These are not commonly used codes and you
may wish not to count words containing them,
just noting them in the count of 'Other'
words.
-reverse boolean [N] Set this to be true if you also wish to
also count words in the reverse complement
of a nucleic sequence.
-calcfreq boolean [N] If this is set true then the expected
frequencies of words are calculated from the
observed frequency of single bases or
residues in the sequences.
If you are reporting a word size of 1
(single bases or residues) then there is no
point in using this option because the
calculated expected frequency will be equal
to the observed frequency.
Calculating the expected frequencies like
this will give an approximation of the
expected frequencies that you might get by
using an input file of frequencies produced
by a previous run of this program. If an
input file of expected word frequencies has
been specified then the values from that
file will be used instead of this
calculation of expected frequency from the
sequence, even if 'calcfreq' is set to be
true.
-[no]zerocount boolean [Y] You can make the output results file
much smaller if you do not display the words
with a zero count.
-sbegin1 integer Start of each sequence to be used
-send1 integer End of each sequence to be used
-sreverse1 boolean Reverse (if DNA)
-snucleotide1 boolean Sequence is nucleotide
-sprotein1 boolean Sequence is protein
-slower1 boolean Make lower case
-supper1 boolean Make upper case
-scircular1 boolean Sequence is circular
Output format:[Contig]\t[Word]\t[Obs Count]\t[Obs Frequency]\t[Exp Frequency]\t[Obs/Exp Frequency] Manual
Example usage:
$ echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./compseq.sh | head -10
NODE_3_length_3390_cov_20.385250 AA 30 0.0086881 0.0625000 0.1390096
NODE_3_length_3390_cov_20.385250 AC 199 0.0576310 0.0625000 0.9220967
NODE_3_length_3390_cov_20.385250 AG 161 0.0466261 0.0625000 0.7460180
NODE_3_length_3390_cov_20.385250 AT 65 0.0188242 0.0625000 0.3011874
NODE_3_length_3390_cov_20.385250 CA 170 0.0492326 0.0625000 0.7877208
NODE_3_length_3390_cov_20.385250 CC 365 0.1057052 0.0625000 1.6912829
NODE_3_length_3390_cov_20.385250 CG 603 0.1746308 0.0625000 2.7940921
NODE_3_length_3390_cov_20.385250 CT 122 0.0353316 0.0625000 0.5653055
NODE_3_length_3390_cov_20.385250 GA 227 0.0657399 0.0625000 1.0518390
NODE_3_length_3390_cov_20.385250 GC 453 0.1311903 0.0625000 2.0990443
$ echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./compseq.sh "-word=3" | head -10
NODE_3_length_3390_cov_20.385250 AAA 0 0.0000000 0.0156250 0.0000000
NODE_3_length_3390_cov_20.385250 AAC 18 0.0052144 0.0156250 0.3337196
NODE_3_length_3390_cov_20.385250 AAG 10 0.0028969 0.0156250 0.1853998
NODE_3_length_3390_cov_20.385250 AAT 2 0.0005794 0.0156250 0.0370800
NODE_3_length_3390_cov_20.385250 ACA 20 0.0057937 0.0156250 0.3707995
NODE_3_length_3390_cov_20.385250 ACC 76 0.0220162 0.0156250 1.4090382
NODE_3_length_3390_cov_20.385250 ACG 93 0.0269409 0.0156250 1.7242178
NODE_3_length_3390_cov_20.385250 ACT 10 0.0028969 0.0156250 0.1853998
NODE_3_length_3390_cov_20.385250 AGA 15 0.0043453 0.0156250 0.2780997
NODE_3_length_3390_cov_20.385250 AGC 82 0.0237543 0.0156250 1.5202781
For drawing the Obs Frequency for a given contig, we can simply use:
$ gplot -H 1 -x "Obs Frequency" 'using 1:3:xtic(2) w boxes lc rgb "green"' ::: <(echo "NODE_3_length_3390_cov_20.385250" | FASTAgrep --buffer-size=100000000 contigs.fa | ./compseq.sh | perl -alne 'print $.."\t".join("\t",@F[1,3])')
to produce: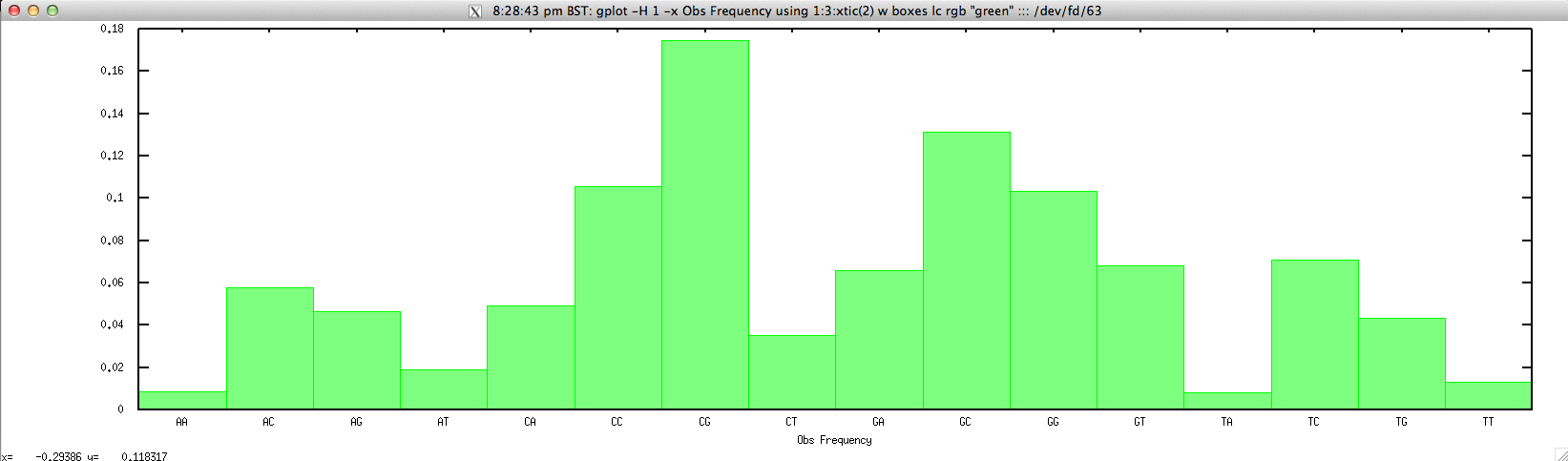
We can also use
GENERATEtable.sh script to compare 3-MERs Obs Frequencey for selected contigs:
$ echo "NODE_14372\d_length_\d+_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./compseq.sh "-word=3" | perl -alne 'print join("\t",@F[0,1,3])' | ./GENERATEtable.sh
Contigs NODE_143720_length_167_cov_3.089820 NODE_143722_length_151_cov_2.304636 NODE_143725_length_172_cov_2.023256 NODE_143728_length_158_cov_2.069620 NODE_143729_length_163_cov_2.128834
AAA 0.1004367 0.0140845 0.0213675 0.0181818 0.0044444
AAC 0.0218341 0.0093897 0.0299145 0.0045455 0.0088889
AAG 0.0174672 0.0140845 0.0170940 0.0136364 0.0088889
AAT 0.0262009 0.0000000 0.0042735 0.0318182 0.0266667
ACA 0.0218341 0.0093897 0.0213675 0.0000000 0.0133333
ACC 0.0087336 0.0140845 0.0170940 0.0090909 0.0222222
ACG 0.0043668 0.0140845 0.0042735 0.0045455 0.0044444
ACT 0.0087336 0.0046948 0.0683761 0.0045455 0.0044444
AGA 0.0218341 0.0093897 0.0042735 0.0090909 0.0177778
AGC 0.0000000 0.0281690 0.0213675 0.0272727 0.0133333
AGG 0.0087336 0.0140845 0.0128205 0.0136364 0.0133333
AGT 0.0393013 0.0093897 0.0170940 0.0090909 0.0222222
ATA 0.0218341 0.0046948 0.0128205 0.0090909 0.0133333
ATC 0.0087336 0.0328638 0.0042735 0.0181818 0.0088889
ATG 0.0131004 0.0187793 0.0085470 0.0136364 0.0266667
ATT 0.0087336 0.0000000 0.0042735 0.0227273 0.0177778
CAA 0.0218341 0.0140845 0.0170940 0.0181818 0.0266667
CAC 0.0000000 0.0187793 0.0256410 0.0000000 0.0133333
CAG 0.0305677 0.0281690 0.0085470 0.0136364 0.0444444
CAT 0.0131004 0.0093897 0.0042735 0.0045455 0.0266667
CCA 0.0131004 0.0281690 0.0170940 0.0000000 0.0355556
CCC 0.0087336 0.0328638 0.0128205 0.0136364 0.0088889
CCG 0.0000000 0.0234742 0.0042735 0.0181818 0.0133333
CCT 0.0349345 0.0281690 0.0213675 0.0181818 0.0133333
CGA 0.0043668 0.0187793 0.0085470 0.0318182 0.0088889
CGC 0.0000000 0.0375587 0.0085470 0.0136364 0.0222222
CGG 0.0000000 0.0234742 0.0000000 0.0318182 0.0133333
CGT 0.0000000 0.0093897 0.0000000 0.0136364 0.0133333
CTA 0.0000000 0.0093897 0.0470085 0.0045455 0.0000000
CTC 0.0087336 0.0234742 0.0427350 0.0045455 0.0222222
CTG 0.0218341 0.0375587 0.0256410 0.0227273 0.0177778
CTT 0.0305677 0.0093897 0.0470085 0.0136364 0.0088889
GAA 0.0218341 0.0093897 0.0256410 0.0227273 0.0088889
GAC 0.0131004 0.0093897 0.0256410 0.0136364 0.0133333
GAG 0.0174672 0.0187793 0.0042735 0.0181818 0.0088889
GAT 0.0043668 0.0375587 0.0128205 0.0181818 0.0133333
GCA 0.0131004 0.0046948 0.0042735 0.0181818 0.0311111
GCC 0.0043668 0.0422535 0.0128205 0.0272727 0.0222222
GCG 0.0000000 0.0328638 0.0000000 0.0500000 0.0222222
GCT 0.0043668 0.0422535 0.0213675 0.0045455 0.0222222
GGA 0.0087336 0.0140845 0.0256410 0.0181818 0.0088889
GGC 0.0000000 0.0328638 0.0085470 0.0136364 0.0266667
GGG 0.0043668 0.0046948 0.0000000 0.0000000 0.0000000
GGT 0.0174672 0.0046948 0.0000000 0.0181818 0.0044444
GTA 0.0131004 0.0000000 0.0085470 0.0090909 0.0088889
GTC 0.0218341 0.0093897 0.0042735 0.0181818 0.0266667
GTG 0.0218341 0.0093897 0.0000000 0.0318182 0.0133333
GTT 0.0218341 0.0046948 0.0042735 0.0136364 0.0133333
TAA 0.0218341 0.0000000 0.0128205 0.0090909 0.0088889
TAC 0.0043668 0.0046948 0.0299145 0.0000000 0.0044444
TAG 0.0087336 0.0000000 0.0256410 0.0136364 0.0044444
TAT 0.0087336 0.0093897 0.0085470 0.0090909 0.0000000
TCA 0.0174672 0.0281690 0.0085470 0.0181818 0.0311111
TCC 0.0349345 0.0234742 0.0128205 0.0045455 0.0177778
TCG 0.0000000 0.0187793 0.0085470 0.0181818 0.0177778
TCT 0.0131004 0.0046948 0.0512821 0.0181818 0.0088889
TGA 0.0218341 0.0328638 0.0299145 0.0136364 0.0088889
TGC 0.0218341 0.0234742 0.0000000 0.0409091 0.0355556
TGG 0.0174672 0.0140845 0.0213675 0.0045455 0.0133333
TGT 0.0218341 0.0000000 0.0000000 0.0318182 0.0222222
TTA 0.0087336 0.0000000 0.0085470 0.0090909 0.0000000
TTC 0.0262009 0.0093897 0.0299145 0.0181818 0.0177778
TTG 0.0262009 0.0046948 0.0170940 0.0227273 0.0222222
TTT 0.0393013 0.0000000 0.0170940 0.0409091 0.0266667
where perl -alne 'print join("\t",@F[0,1,3])' extracts data in the right format for GENERATEtable.sh.
dan.sh: calculate nucleic acid melting temperature
Content of dan.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/dan -sequence=asis:$a[1] $o -stdout -auto 2>\/dev\/null/;
$r=~s/#.*\n//g;
$r=~s/\s+\n//g;
$r=~s/\s+Start.*?\n//g;
$r=~s/^\s+//g;
$r=~s/\n\s+/\n/g;
$r=~s/\h+/\t/g;
if (defined $r and length $r){
print substr($a[0],1)."\t".join("\n".substr($a[0],1)."\t",split("\n",$r))."\n"}}' -- -o=$1
Parameters [from dan -help -verbose]:
-windowsize integer [20] The values of melting point and other
thermodynamic properties of the sequence are
determined by taking a short length of
sequence known as a window and determining
the properties of the sequence in that
window. The window is incrementally moved
along the sequence with the properties being
calculated at each new position. (Integer
from 1 to 100)
-shiftincrement integer [1] This is the amount by which the window
is moved at each increment in order to find
the melting point and other properties along
the sequence. (Integer 1 or more)
-dnaconc float [50.] Enter DNA concentration (nM) (Number
from 1.000 to 100000.000)
-saltconc float [50.] Enter salt concentration (mM) (Number
from 1.000 to 1000.000)
-mintemp float [55.] Enter a minimum value for the
temperature scale (y-axis) of the plot.
(Number from 0.000 to 150.000)
-product toggle This prompts for percent formamide, percent
of mismatches allowed and product length.
-formamide float [0.] This specifies the percent formamide to
be used in calculations (it is ignored
unless -product is used). (Number from 0.000
to 100.000)
-mismatch float [0.] This specifies the percent mismatch to
be used in calculations (it is ignored
unless -product is used). (Number from 0.000
to 100.000)
-prodlen integer [Window size (20)] This specifies the
product length to be used in calculations
(it is ignored unless -product is used).
(Any integer value)
-thermo toggle Output the DeltaG, DeltaH and DeltaS values
of the sequence windows to the output data
file.
-temperature float [25.] If -thermo has been specified then
this specifies the temperature at which to
calculate the DeltaG, DeltaH and DeltaS
values. (Number from 0.000 to 100.000)
-rna boolean This specifies that the sequence is an RNA
sequence and not a DNA sequence.
-sbegin1 integer Start of each sequence to be used
-send1 integer End of each sequence to be used
-sreverse1 boolean Reverse (if DNA)
-snucleotide1 boolean Sequence is nucleotide
-sprotein1 boolean Sequence is protein
-slower1 boolean Make lower case
-supper1 boolean Make upper case
-scircular1 boolean Sequence is circular
Output format:[Contig]\t[Start]\t[End]\t[Strand]\t[Tm]\t[GC]\t[DeltaG]\t[DeltaH]\t[DeltaS]\t[TmProd]\t[Sequence] Manual
Example usage:
$ echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./dan.sh "-thermo"| head -10
NODE_3_length_3390_cov_20.385250 1 20 + 70.8 80.0 -39.486 -171.000 -441.100 . GCGGGCTCGTTGCCCGCGAC
NODE_3_length_3390_cov_20.385250 2 21 + 70.5 80.0 -39.451 -171.800 -443.900 . CGGGCTCGTTGCCCGCGACG
NODE_3_length_3390_cov_20.385250 3 22 + 70.3 80.0 -39.028 -169.200 -436.600 . GGGCTCGTTGCCCGCGACGG
NODE_3_length_3390_cov_20.385250 4 23 + 70.8 80.0 -39.486 -171.000 -441.100 . GGCTCGTTGCCCGCGACGGC
NODE_3_length_3390_cov_20.385250 5 24 + 70.8 80.0 -39.486 -171.000 -441.100 . GCTCGTTGCCCGCGACGGCC
NODE_3_length_3390_cov_20.385250 6 25 + 69.1 75.0 -38.500 -169.000 -437.700 . CTCGTTGCCCGCGACGGCCT
NODE_3_length_3390_cov_20.385250 7 26 + 69.1 75.0 -38.542 -169.400 -438.900 . TCGTTGCCCGCGACGGCCTC
NODE_3_length_3390_cov_20.385250 8 27 + 70.5 80.0 -39.451 -171.800 -443.900 . CGTTGCCCGCGACGGCCTCG
NODE_3_length_3390_cov_20.385250 9 28 + 70.3 80.0 -39.028 -169.200 -436.600 . GTTGCCCGCGACGGCCTCGG
NODE_3_length_3390_cov_20.385250 10 29 + 70.3 75.0 -39.028 -169.200 -436.600 . TTGCCCGCGACGGCCTCGGT
The graph for Tm is given below:
$ gplot -x "Tm" 'using 1:2 w l' ::: <(echo "NODE_3_length_3390_cov_20.385250" | FASTAgrep --buffer-size=100000000 contigs.fa | ./dan.sh "-thermo" | perl -alne 'print join("\t",@F[1,4])')
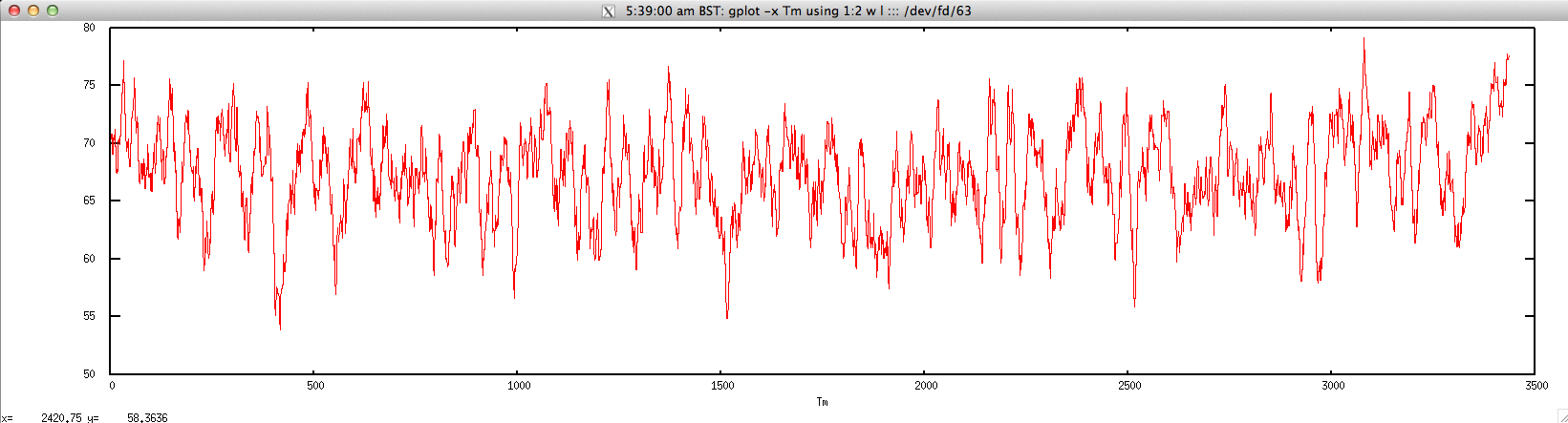
density.sh: calculate nucleic acid density
Content of density.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/density -seqall=asis:$a[1] $o -stdout -auto 2>\/dev\/null/;
$r=~s/#.*\n//g;
$r=~s/\s+\n//g;
$r=~s/\s+Start.*?\n//g;
$r=~s/^\s+//g;
$r=~s/\n\s+/\n/g;
$r=~s/\h+/\t/g;
if (defined $r and length $r){
print substr($a[0],1)."\t".join("\n".substr($a[0],1)."\t",split("\n",$r))."\n"}}' -- -o=$1
Parameters [from density -help -verbose]:
-window integer [100] Window length (Integer 1 or more) -sbegin1 integer Start of each sequence to be used -send1 integer End of each sequence to be used -sreverse1 boolean Reverse (if DNA) -sprotein1 boolean Sequence is protein -slower1 boolean Make lower case -supper1 boolean Make upper case -scircular1 boolean Sequence is circularOutput format:
[Contig]\t[Start]\t[End]\t[Strand]\t[Score]\t[a]\t[c]\t[g]\t[t]\t[at]\t[gc] Manual
Example usage:
$ echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./density.sh | head -10
NODE_3_length_3390_cov_20.385250 1 1 + 0.000 0.100 0.370 0.420 0.110 0.210 0.790
NODE_3_length_3390_cov_20.385250 2 2 + 0.000 0.100 0.380 0.410 0.110 0.210 0.790
NODE_3_length_3390_cov_20.385250 3 3 + 0.000 0.100 0.370 0.420 0.110 0.210 0.790
NODE_3_length_3390_cov_20.385250 4 4 + 0.000 0.100 0.370 0.410 0.120 0.220 0.780
NODE_3_length_3390_cov_20.385250 5 5 + 0.000 0.100 0.380 0.400 0.120 0.220 0.780
NODE_3_length_3390_cov_20.385250 6 6 + 0.000 0.100 0.380 0.400 0.120 0.220 0.780
NODE_3_length_3390_cov_20.385250 7 7 + 0.000 0.100 0.370 0.400 0.130 0.230 0.770
NODE_3_length_3390_cov_20.385250 8 8 + 0.000 0.100 0.380 0.400 0.120 0.220 0.780
NODE_3_length_3390_cov_20.385250 9 9 + 0.000 0.100 0.370 0.410 0.120 0.220 0.780
NODE_3_length_3390_cov_20.385250 10 10 + 0.000 0.100 0.370 0.410 0.120 0.220 0.780
We can plot them for a given contig as follows:
$ echo "NODE_3_length_3390_cov_20.385250" | FASTAgrep --buffer-size=100000000 contigs.fa | ./density.sh | \
perl -alne 'print join("\t",@F[1,5,6,7,8,9,10])' > per_base_SQ.dat; \
gplot -e -x "Sequence content across all bases" u 1:2 t \"a\" w l, \"\" u 1:3 t \"c\" w l, \"\" u 1:4 t \"g\" w l, \"\" u 1:5 t \"t\" w l, \"\" u 1:6 t \"at\" w l, \"\" u 1:7 t \"gc\" w l ::: per_base_SQ.dat; rm per_base_SQ.dat
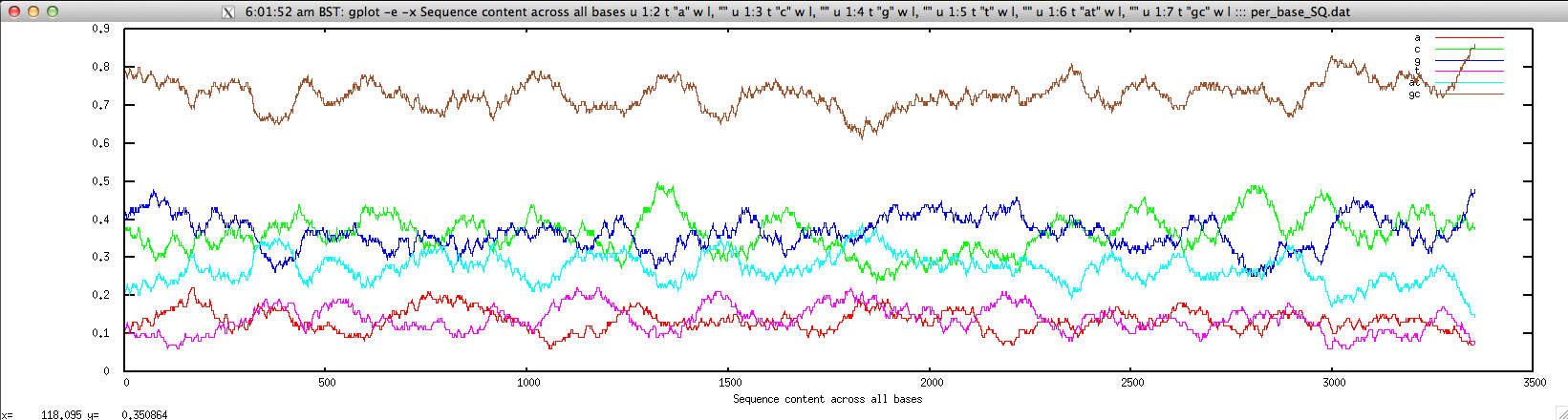
cpgreport.sh: identify and report CpG-rich regions in nucleotide sequence
Content of cpgreport.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/cpgreport -sequence=asis:$a[1] $o -stdout -auto 2>\/dev\/null/;
$r=~s/#.*\n//g;
$r=~s/\s*\n\s*/\n/g;
$r=~s/CPGREPORT.*?\n//g;
$r=~s/\nSequence.*?\n//g;
$r=~s/asis\s+//g;
$r=~s/\h+/\t/g;
if (defined $r and length $r){
print substr($a[0],1)."\t".join("\n".substr($a[0],1)."\t",split("\n",$r))."\n"}}' -- -o=$1
Parameters [from cpgreport -help -verbose]):
-score integer [17] This sets the score for each CG
sequence found. A value of 17 is more
sensitive, but 28 has also been used with
some success. (Integer from 1 to 200)
-sbegin1 integer Start of each sequence to be used
-send1 integer End of each sequence to be used
-sreverse1 boolean Reverse (if DNA)
-snucleotide1 boolean Sequence is nucleotide
-sprotein1 boolean Sequence is protein
-slower1 boolean Make lower case
-supper1 boolean Make upper case
-scircular1 boolean Sequence is circular
Output format:[Contig]\t[Begin]\t[End]\t[Score]\t[CpG]\t[%CG]\t[CG/GC] Manual
Example usage:
$ echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./cpgreport.sh | head -10
NODE_3_length_3390_cov_20.385250 2 3452 7404 603 73.3 1.34
NODE_6_length_5675_cov_18.628546 1 5739 11380 951 73.2 1.20
NODE_11_length_1365_cov_14.082784 6 1424 3424 269 76.9 1.27
NODE_12_length_1944_cov_7.141975 5 1995 4292 349 71.9 1.54
NODE_13_length_1418_cov_18.503527 1 1480 2355 213 71.2 1.13
NODE_17_length_5129_cov_24.638330 5 5190 10835 890 73.9 1.21
NODE_18_length_2905_cov_18.701204 14 2966 5670 479 71.4 1.24
NODE_20_length_6239_cov_17.427153 2 6302 14022 1129 75.3 1.27
NODE_22_length_4091_cov_19.720362 3 4155 8250 689 73.3 1.21
NODE_24_length_4513_cov_14.317084 6 4575 7041 645 68.9 1.21
To graphically display No. of CpGs in the given range, use
$ gplot -H 1 -x "No. of CpGs" 'using 1:2 w boxes lc rgb "blue"' ::: <(echo "NODE_3_length_3390_cov_20.385250" | FASTAgrep --buffer-size=100000000 contigs.fa | \
./cpgreport.sh -score="5" | \
perl -alne 'for($i=$F[1];$i<=$F[2];$i++){print $i."\t".$F[3]}')
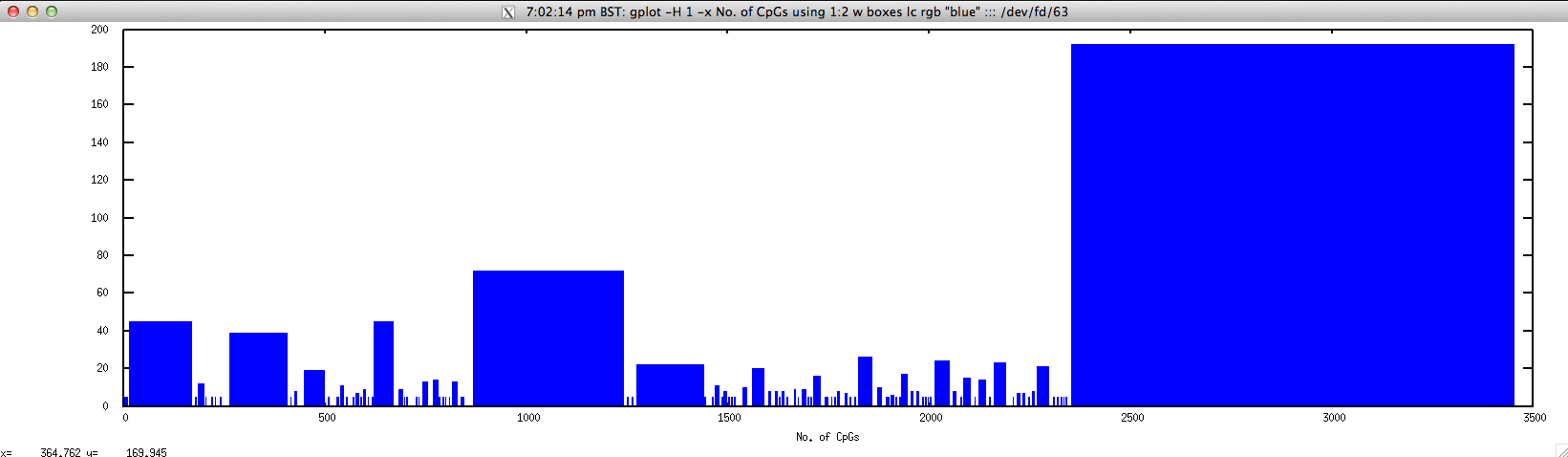
newcpgreport.sh: identify CpG islands in nucleotide sequence
Content of newcpgreport.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/newcpgreport -sequence=asis:$a[1] $o -stdout -auto 2>\/dev\/null/;
$r=~s/^(.*?\n)+?FT/FT/g;
$r=~s/FT\s+CpG island\s+(\d+)\.\.(\d+)\n/\n\1\t\2\t/g;
$r=~s/FT\s+\/size=(\d+)\n/\1\t/g;
$r=~s/FT\s+\/Sum C\+G=(\d+)\n/\1\t/g;
$r=~s/FT\s+\/Percent CG=(\d+\.\d+)\n/\1\t/g;
$r=~s/FT\s+\/ObsExp=(\d+\.\d+)\n/\1/g;
$r=~s/FT\s+numislands\s+\d+\s*\n\/\///g;
$r=~s/^\n//g;
if (defined $r and length $r){
print substr($a[0],1)."\t".join("\n".substr($a[0],1)."\t",split("\n",$r))."\n"}}' -- -o=$1
Parameters [from newcpgreport -help -verbose]:
-window integer [100] Window size (Integer 1 or more)
-shift integer [1] Shift increment (Integer 1 or more)
-minlen integer [200] Minimum Length (Integer 1 or more)
-minoe float [0.6] Minimum observed/expected (Number from
0.000 to 10.000)
-minpc float [50.] Minimum percentage (Number from 0.000
to 100.000)
-sbegin1 integer Start of each sequence to be used
-send1 integer End of each sequence to be used
-sreverse1 boolean Reverse (if DNA)
-snucleotide1 boolean Sequence is nucleotide
-sprotein1 boolean Sequence is protein
-slower1 boolean Make lower case
-supper1 boolean Make upper case
-scircular1 boolean Sequence is circular
Output format:[Contig]\t[Begin]\t[End]\t[Size]\t[Sum C+G]\t[Percent CG]\t[ObsExp] Manual
Example usage:
$ echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./newcpgreport.sh | head -10
NODE_3_length_3390_cov_20.385250 47 3400 3354 2444 72.87 1.30
NODE_6_length_5675_cov_18.628546 50 5684 5635 4122 73.15 1.24
NODE_11_length_1365_cov_14.082784 48 1374 1327 1025 77.24 1.27
NODE_12_length_1944_cov_7.141975 47 1952 1906 1370 71.88 1.36
NODE_13_length_1418_cov_18.503527 48 1426 1379 985 71.43 1.13
NODE_17_length_5129_cov_24.638330 47 5139 5093 3762 73.87 1.26
NODE_18_length_2905_cov_18.701204 47 2913 2867 2041 71.19 1.27
NODE_20_length_6239_cov_17.427153 47 6249 6203 4666 75.22 1.26
NODE_22_length_4091_cov_19.720362 47 4101 4055 2959 72.97 1.24
NODE_24_length_4513_cov_14.317084 47 4521 4475 3081 68.85 1.18
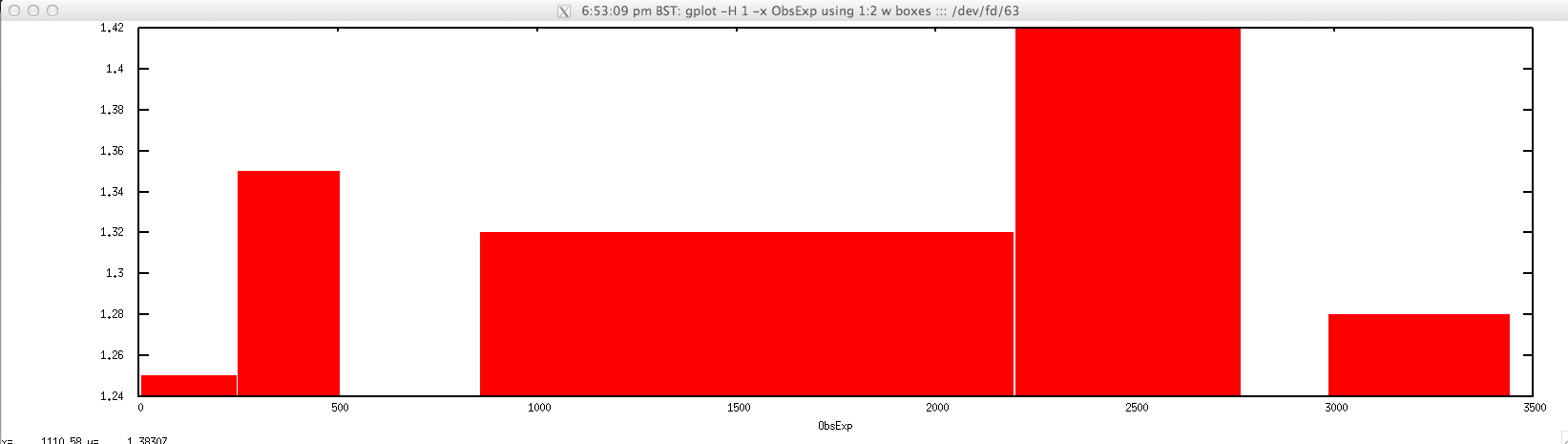
For plotting ObsExp for a selected contig:
$ gplot -H 1 -x "ObsExp" 'using 1:2 w boxes' ::: <(echo "NODE_3_length_3390_cov_20.385250" | \
FASTAgrep --buffer-size=100000000 contigs.fa | ./newcpgreport.sh "-window=20" | \
perl -alne 'for($i=$F[1];$i<=$F[2];$i++){print $i."\t".$F[6]}')
fuzznuc.sh: search for patterns in nucleotide sequences
Content of fuzznuc.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/fuzznuc -sequence=asis:$a[1] $o -stdout -auto 2>\/dev\/null/;
$r=~s/#.*\n//g;
$r=~s/\s+\n//g;
$r=~s/\s+Start.*?\n//g;
$r=~s/^\s+//g;
$r=~s/\n\s+/\n/g;
$r=~s/\h+/\t/g;
if (defined $r and length $r){
print substr($a[0],1)."\t".join("\n".substr($a[0],1)."\t",split("\n",$r))."\n"}}' -- -o=$1
Parameters [from fuzznuc -help -verbose]:
-pattern pattern The standard IUPAC one-letter codes for the
nucleotides are used.
The symbol 'n' is used for a position where
any nucleotide is accepted.
Ambiguities are indicated by listing the
acceptable nucleotides for a given position,
between square parentheses '[ ]'. For
example: [ACG] stands for A or C or G.
Ambiguities are also indicated by listing
between a pair of curly brackets '{ }' the
nucleotides that are not accepted at a given
position. For example: {AG} stands for any
nucleotides except A and G.
Each element in a pattern is separated from
its neighbor by a '-'. (Optional in
fuzznuc).
Repetition of an element of the pattern can
be indicated by following that element with
a numerical value or a numerical range
between parenthesis. Examples: N(3)
corresponds to N-N-N, N(2,4) corresponds to
N-N or N-N-N or N-N-N-N.
When a pattern is restricted to either the
5' or 3' end of a sequence, that pattern
either starts with a '<' symbol or
respectively ends with a '>' symbol.
A period ends the pattern. (Optional in
fuzznuc).
For example, [CG](5)TG{A}N(1,5)C
-complement boolean [N] Search complementary strand
-sbegin1 integer Start of each sequence to be used
-send1 integer End of each sequence to be used
-sreverse1 boolean Reverse (if DNA)
-snucleotide1 boolean Sequence is nucleotide
-sprotein1 boolean Sequence is protein
-slower1 boolean Make lower case
-supper1 boolean Make upper case
-scircular1 boolean Sequence is circular
-pmismatch integer Pattern mismatch
-pname string Pattern base name
Output format:
[Contig]\t[Start]\t[End]\t[Strand]\t[Pattern]\t[Mismatch]\t[Sequence] Manual
Example usage:
$ echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./fuzznuc.sh "-pattern=[CG]\(5\)TG{A}N\(1,5\)C" | head -10
NODE_3_length_3390_cov_20.385250 3424 3433 + pattern:[CG](5)TG{A}N(1,5)C . CGGCCTGCGC
NODE_3_length_3390_cov_20.385250 3424 3436 + pattern:[CG](5)TG{A}N(1,5)C . CGGCCTGCGCGGC
NODE_3_length_3390_cov_20.385250 3349 3361 + pattern:[CG](5)TG{A}N(1,5)C . CGCGCTGGGGTTC
NODE_3_length_3390_cov_20.385250 3001 3010 + pattern:[CG](5)TG{A}N(1,5)C . GCGGCTGCCC
NODE_3_length_3390_cov_20.385250 3001 3011 + pattern:[CG](5)TG{A}N(1,5)C . GCGGCTGCCCC
NODE_3_length_3390_cov_20.385250 3001 3013 + pattern:[CG](5)TG{A}N(1,5)C . GCGGCTGCCCCGC
NODE_3_length_3390_cov_20.385250 2914 2923 + pattern:[CG](5)TG{A}N(1,5)C . CGCCCTGCGC
NODE_3_length_3390_cov_20.385250 2914 2924 + pattern:[CG](5)TG{A}N(1,5)C . CGCCCTGCGCC
NODE_3_length_3390_cov_20.385250 2914 2926 + pattern:[CG](5)TG{A}N(1,5)C . CGCCCTGCGCCAC
NODE_3_length_3390_cov_20.385250 2568 2580 + pattern:[CG](5)TG{A}N(1,5)C . CGCCGTGGCGGAC
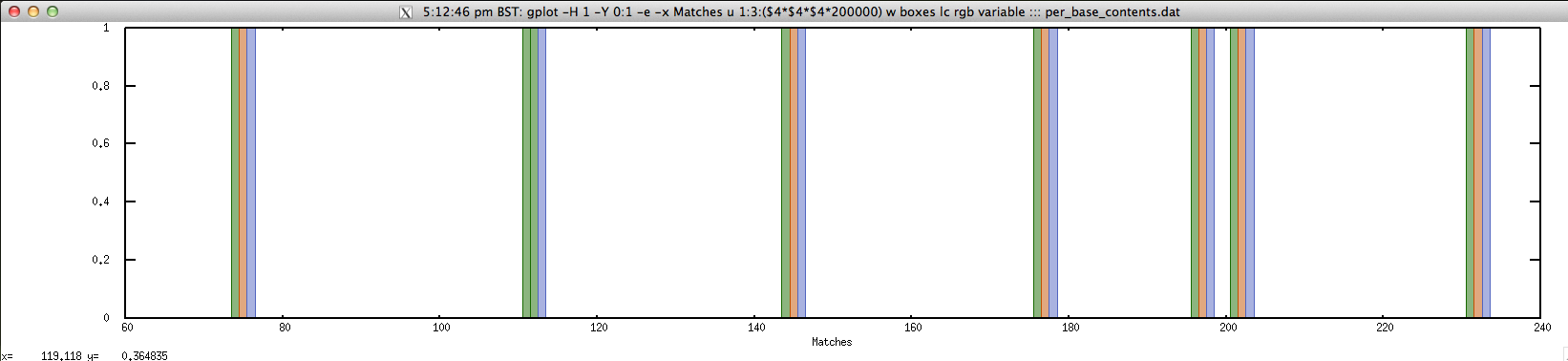
For shorter contigs, we can view the matches on the length of the contig:
$ echo "NODE_143725_length_172_cov_2.023256" | FASTAgrep --buffer-size=100000000 contigs.fa | \
./fuzznuc.sh "-pattern=C{A}G" | \
perl -ane '$j=0;@v=split("",$F[6]);for($i=$F[1];$i<=$F[2];$i++){$r{$i}=$v[$j++]}}{foreach my $n (sort { $a <=> $b} keys %r){print $n."\t".$r{$n};$r{$n}=~tr/ACGTRYSWKMBDHVN-/1234555555555555/;print "\t1\t".$r{$n}."\n"}' > per_base_contents.dat
$ cat per_base_content.dat | head -10
74 C 1 2
75 T 1 4
76 G 1 3
111 C 1 2
112 C 1 2
113 G 1 3
144 C 1 2
145 T 1 4
146 G 1 3
176 C 1 2
$ gplot -H 1 -Y 0:1 -e -x "Matches" 'u 1:3:($4*$4*$4*200000) w boxes lc rgb variable' ::: per_base_content.dat; rm per_base_content.dat
fuzztran.sh: search for patterns in protein sequences (translated)
Content of fuzztran.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/fuzztran -sequence=asis:$a[1] $o -stdout -auto 2>\/dev\/null/;
$r=~s/#.*\n//g;
$r=~s/\s*\n\s*/\n/g;
$r=~s/\nStart.*?\n//g;
$r=~s/^\s+//g;
$r=~s/\n\s+/\n/g;
$r=~s/\h+/\t/g;
if (defined $r and length $r){
print substr($a[0],1)."\t".join("\n".substr($a[0],1)."\t",split("\n",$r))."\n"}}' -- -o=$1
Parameters [from fuzztran -help -verbose]:
-pattern pattern The standard IUPAC one-letter codes for the
amino acids are used.
The symbol 'x' is used for a position where
any amino acid is accepted.
Ambiguities are indicated by listing the
acceptable amino acids for a given position,
between square parentheses '[ ]'. For
example: [ALT] stands for Ala or Leu or Thr.
Ambiguities are also indicated by listing
between a pair of curly brackets '{ }' the
amino acids that are not accepted at a gven
position. For example: {AM} stands for any
amino acid except Ala and Met.
Each element in a pattern is separated from
its neighbor by a '-'. (Optional in
fuzztran)
Repetition of an element of the pattern can
be indicated by following that element with
a numerical value or a numerical range
between parenthesis. Examples: x(3)
corresponds to x-x-x, x(2,4) corresponds to
x-x or x-x-x or x-x-x-x.
When a pattern is restricted to either the
N- or C-terminal of a sequence, that pattern
either starts with a '<' symbol or
respectively ends with a '>' symbol.
A period ends the pattern. (Optional in
fuzztran).
-frame menu [1] Frame(s) to translate (Values: 1 (1); 2
(2); 3 (3); F (Forward three frames); -1
(-1); -2 (-2); -3 (-3); R (Reverse three
frames); 6 (All six frames))
-table menu [0] Code to use (Values: 0 (Standard); 1
(Standard (with alternative initiation
codons)); 2 (Vertebrate Mitochondrial); 3
(Yeast Mitochondrial); 4 (Mold, Protozoan,
Coelenterate Mitochondrial and
Mycoplasma/Spiroplasma); 5 (Invertebrate
Mitochondrial); 6 (Ciliate Macronuclear and
Dasycladacean); 9 (Echinoderm
Mitochondrial); 10 (Euplotid Nuclear); 11
(Bacterial); 12 (Alternative Yeast Nuclear);
13 (Ascidian Mitochondrial); 14 (Flatworm
Mitochondrial); 15 (Blepharisma
Macronuclear); 16 (Chlorophycean
Mitochondrial); 21 (Trematode
Mitochondrial); 22 (Scenedesmus obliquus);
23 (Thraustochytrium Mitochondrial))
-sbegin1 integer Start of each sequence to be used
-send1 integer End of each sequence to be used
-sreverse1 boolean Reverse (if DNA)
-snucleotide1 boolean Sequence is nucleotide
-sprotein1 boolean Sequence is protein
-slower1 boolean Make lower case
-supper1 boolean Make upper case
-scircular1 boolean Sequence is circular
-pmismatch integer Pattern mismatch
-pname string Pattern base name
Output format:[Contig]\t[Start]\t[End]\t[Strand]\t[Score]\t[Pattern]\t[Mismatch]\t[Frame]\t[PStart]\t[PEnd]\t[Translation] Manual
Example usage:
$ echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./fuzztran.sh "-pattern=V{V}VVVL" | head -10
NODE_31_length_15435_cov_19.081308 13504 13521 + 6 pattern:V{V}VVVL . 1 4502 4507 VTVVVL
NODE_729_length_5451_cov_18.101265 4561 4578 + 6 pattern:V{V}VVVL . 1 1521 1526 VAVVVL
NODE_934_length_8261_cov_16.588669 5125 5142 + 6 pattern:V{V}VVVL . 1 1709 1714 VGVVVL
NODE_1805_length_3627_cov_17.087124 1396 1413 + 6 pattern:V{V}VVVL . 1 466 471 VGVVVL
NODE_2046_length_9730_cov_19.413464 9202 9219 + 6 pattern:V{V}VVVL . 1 3068 3073 VLVVVL
NODE_2514_length_8896_cov_14.840940 7588 7605 + 6 pattern:V{V}VVVL . 1 2530 2535 VLVVVL
NODE_5074_length_2208_cov_10.750453 619 636 + 6 pattern:V{V}VVVL . 1 207 212 VTVVVL
NODE_8554_length_1025_cov_11.535610 847 864 + 6 pattern:V{V}VVVL . 1 283 288 VDVVVL
NODE_8554_length_1025_cov_11.535610 922 939 + 6 pattern:V{V}VVVL . 1 308 313 VAVVVL
NODE_10591_length_4696_cov_18.995316 3610 3627 + 6 pattern:V{V}VVVL . 1 1204 1209 VTVVVL
freak.sh: generate residue/base frequency table
Content of freak.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/freak -seqall=asis:$a[1] $o -stdout -auto 2>\/dev\/null/;
$r=~s/\s+\n/\n/g;
$r=~s/FREAK.*?\n//g;
$r=~s/^\s+//g;
$r=~s/\n\s+/\n/g;
$r=~s/\h+/\t/g;
if (defined $r and length $r){
print substr($a[0],1)."\t".join("\n".substr($a[0],1)."\t",split("\n",$r))."\n"}}' -- -o=$1
Parameters [from freak -help -verbose]:
-letters string [gc] Residue letters (Any string) -step integer [1] Stepping value (Any integer value) -window integer [30] Averaging window (Any integer value) -sbegin1 integer Start of each sequence to be used -send1 integer End of each sequence to be used -sreverse1 boolean Reverse (if DNA) -snucleotide1 boolean Sequence is nucleotide -sprotein1 boolean Sequence is protein -slower1 boolean Make lower case -supper1 boolean Make upper case -scircular1 boolean Sequence is circularOutput format:
[Contig]\t[Start]\t[Frequency] Manual
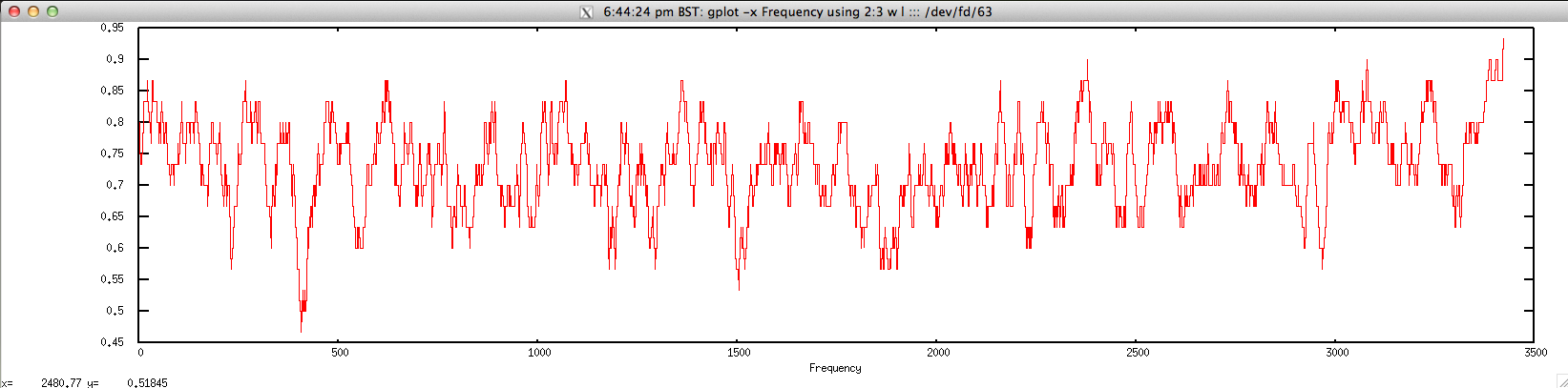
Example usage:
$ echo "NODE_3_length_3390_cov_20.385250" | FASTAgrep --buffer-size=100000000 contigs.fa | ./freak.sh | head -10
NODE_3_length_3390_cov_20.385250 1 0.800000
NODE_3_length_3390_cov_20.385250 2 0.766667
NODE_3_length_3390_cov_20.385250 3 0.733333
NODE_3_length_3390_cov_20.385250 4 0.733333
NODE_3_length_3390_cov_20.385250 5 0.733333
NODE_3_length_3390_cov_20.385250 6 0.733333
NODE_3_length_3390_cov_20.385250 7 0.733333
NODE_3_length_3390_cov_20.385250 8 0.766667
NODE_3_length_3390_cov_20.385250 9 0.766667
NODE_3_length_3390_cov_20.385250 10 0.766667
$ gplot -x "Frequency" 'using 2:3 w l' ::: <(echo "NODE_3_length_3390_cov_20.385250" | FASTAgrep --buffer-size=100000000 contigs.fa | ./freak.sh)
etandem.sh: find tandem repeats in a nucleotide sequence
Content of etandem.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/etandem -sequence=asis:$a[1] $o -stdout -auto 2>\/dev\/null/;
$r=~s/#.*\n//g;
$r=~s/\s+Start.*?\n//g;
$r=~s/^\s+//g;
$r=~s/\n\s+/\n/g;
$r=~s/\h+/\t/g;
if (defined $r and length $r){
print substr($a[0],1)."\t".join("\n".substr($a[0],1)."\t",split("\n",$r))."\n"}}' -- -o=$1
Parameters [from etandem -help -verbose]:
-minrepeat integer [10] Minimum repeat size (Integer, 2 or
higher)
-maxrepeat integer [Same as -minrepeat] Maximum repeat size
(Integer, same as -minrepeat or higher)
-threshold integer [20] Threshold score (Any integer value)
-mismatch boolean Allow N as a mismatch
-uniform boolean Allow uniform consensus
-sbegin1 integer Start of the sequence to be used
-send1 integer End of the sequence to be used
-sreverse1 boolean Reverse (if DNA)
-snucleotide1 boolean Sequence is nucleotide
-sprotein1 boolean Sequence is protein
-slower1 boolean Make lower case
-supper1 boolean Make upper case
-scircular1 boolean Sequence is circular
Output format:[Contig]\t[Start]\t[End]\t[Strand]\t[Score]\t[Size]\t[Count]\t[Identity]\t[Consensus] Manual
Example usage:
$ echo "NODE_\d+_length_(\d){4,}_" | FASTAgrep --buffer-size=100000000 contigs.fa | ./etandem.sh "-minrepeat=5" | head -10
NODE_554_length_5056_cov_16.977057 4517 4576 + 21 5 12 71.7 cgcgg
NODE_644_length_7589_cov_17.300699 5464 5578 + 20 5 23 60.9 ggcag
NODE_1822_length_12545_cov_19.939497 6789 6903 + 38 5 23 68.7 gcccg
NODE_1824_length_12270_cov_17.698696 8624 8698 + 20 5 15 66.7 cgccg
NODE_1825_length_3666_cov_17.308783 570 624 + 20 5 11 72.7 ccgcc
NODE_1860_length_13418_cov_18.133999 616 730 + 20 5 23 60.9 ccgcc
NODE_2108_length_12563_cov_19.594921 1376 1440 + 22 5 13 70.8 ggccg
NODE_2175_length_7718_cov_23.064524 1406 1480 + 24 5 15 69.3 gggcg
NODE_2472_length_2810_cov_16.679716 484 578 + 20 5 19 63.2 cgggg
NODE_2982_length_3833_cov_14.351161 547 726 + 43 5 36 63.3 ggggc
tcode.sh: identify protein-coding regions using Fickett TESTCODE statistic
Content of tcode.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/tcode -sequence=asis:$a[1] $o -stdout -auto 2>\/dev\/null/;
$r=~s/#.*\n//g;
$r=~s/\s+Start.*?\n//g;
$r=~s/No opinion/No_opinion/g;
$r=~s/^\s+//g;
$r=~s/\n\s+/\n/g;
$r=~s/\h+/\t/g;
if (defined $r and length $r){
print substr($a[0],1)."\t".join("\n".substr($a[0],1)."\t",split("\n",$r))."\n"}}' -- -o=$1
Parameters [from tcode -help -verbose]:
-window integer [200] This is the number of nucleotide bases
over which the TESTCODE statistic will be
performed each time. The window will then
slide along the sequence, covering the same
number of bases each time. (Integer 200 or
more)
-step integer [3] The selected window will, by default,
slide along the nucleotide sequence by three
bases at a time, retaining the frame
(although the algorithm is not frame
sensitive). This may be altered to increase
or decrease the increment of the slide.
(Integer 1 or more)
-sbegin1 integer Start of each sequence to be used
-send1 integer End of each sequence to be used
-sreverse1 boolean Reverse (if DNA)
-snucleotide1 boolean Sequence is nucleotide
-sprotein1 boolean Sequence is protein
-slower1 boolean Make lower case
-supper1 boolean Make upper case
-scircular1 boolean Sequence is circular
Output format:[Contig]\t[Start]\t[End]\t[Strand]\t[Score]\t[Estimation] Manual
Example usage:
$ echo "NODE_3_length_3390_cov_20.385250" | FASTAgrep --buffer-size=100000000 contigs.fa | ./tcode.sh | head -10 NODE_3_length_3390_cov_20.385250 1 200 + 1.250 Coding NODE_3_length_3390_cov_20.385250 4 203 + 1.250 Coding NODE_3_length_3390_cov_20.385250 7 206 + 1.250 Coding NODE_3_length_3390_cov_20.385250 10 209 + 1.250 Coding NODE_3_length_3390_cov_20.385250 13 212 + 1.250 Coding NODE_3_length_3390_cov_20.385250 16 215 + 1.250 Coding NODE_3_length_3390_cov_20.385250 19 218 + 1.250 Coding NODE_3_length_3390_cov_20.385250 22 221 + 1.250 Coding NODE_3_length_3390_cov_20.385250 25 224 + 1.232 Coding NODE_3_length_3390_cov_20.385250 28 227 + 1.232 Coding $ echo "NODE_143725_length_172_cov_2.023256" | FASTAgrep --buffer-size=100000000 contigs.fa | ./tcode.sh | head -10 NODE_143725_length_172_cov_2.023256 1 200 + 0.480 Non-coding NODE_143725_length_172_cov_2.023256 4 203 + 0.469 Non-coding NODE_143725_length_172_cov_2.023256 7 206 + 0.556 Non-coding NODE_143725_length_172_cov_2.023256 10 209 + 0.624 Non-coding NODE_143725_length_172_cov_2.023256 13 212 + 0.590 Non-coding NODE_143725_length_172_cov_2.023256 16 215 + 0.590 Non-coding NODE_143725_length_172_cov_2.023256 19 218 + 0.658 Non-coding NODE_143725_length_172_cov_2.023256 22 221 + 0.527 Non-coding NODE_143725_length_172_cov_2.023256 25 224 + 0.576 Non-coding NODE_143725_length_172_cov_2.023256 28 227 + 0.548 Non-coding
getorf.sh: find and extract open reading frames (ORFs)
Content of getorf.sh:
#!/bin/bash
less <&0| \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
chomp $a[1];
$r=qx/getorf -sequence=asis:$a[1] $o -stdout -auto 2>\/dev\/null/;
$r =~ s/>asis/$a[0]/g;
print $r}' -- -o=$2 | \
perl -pe '/^>/?s/^>/\n>/:s/\s*$// if$.>1' | \
perl -nse 'push @a, $_; @a = @a[@a-2..$#a];
if ($. % 2 == 0){
chomp $a[0];
$a[0]=~/>(.*?) \[(\d+) - (\d+)\]\s*(.*)/g;
$s=(($4=~y===c)=="0")?"+":"-";
if($f eq "f"){
print ">".$1."_".$2."_".$3."_".$s."\n".$a[1]}
elsif($f eq "t"){
print $1."\t".$2."\t".$3."\t".$4."\t".$s."\t".$a[1]}}' -- -f=$1
Parameters [from getorf -help -verbose]:
-table menu [0] Code to use (Values: 0 (Standard); 1
(Standard (with alternative initiation
codons)); 2 (Vertebrate Mitochondrial); 3
(Yeast Mitochondrial); 4 (Mold, Protozoan,
Coelenterate Mitochondrial and
Mycoplasma/Spiroplasma); 5 (Invertebrate
Mitochondrial); 6 (Ciliate Macronuclear and
Dasycladacean); 9 (Echinoderm
Mitochondrial); 10 (Euplotid Nuclear); 11
(Bacterial); 12 (Alternative Yeast Nuclear);
13 (Ascidian Mitochondrial); 14 (Flatworm
Mitochondrial); 15 (Blepharisma
Macronuclear); 16 (Chlorophycean
Mitochondrial); 21 (Trematode
Mitochondrial); 22 (Scenedesmus obliquus);
23 (Thraustochytrium Mitochondrial))
-minsize integer [30] Minimum nucleotide size of ORF to
report (Any integer value)
-maxsize integer [1000000] Maximum nucleotide size of ORF to
report (Any integer value)
-find menu [0] This is a small menu of possible output
options. The first four options are to
select either the protein translation or the
original nucleic acid sequence of the open
reading frame. There are two possible
definitions of an open reading frame: it can
either be a region that is free of STOP
codons or a region that begins with a START
codon and ends with a STOP codon. The last
three options are probably only of interest
to people who wish to investigate the
statistical properties of the regions around
potential START or STOP codons. The last
option assumes that ORF lengths are
calculated between two STOP codons. (Values:
0 (Translation of regions between STOP
codons); 1 (Translation of regions between
START and STOP codons); 2 (Nucleic sequences
between STOP codons); 3 (Nucleic sequences
between START and STOP codons); 4
(Nucleotides flanking START codons); 5
(Nucleotides flanking initial STOP codons);
6 (Nucleotides flanking ending STOP codons))
-[no]methionine boolean [Y] START codons at the beginning of protein
products will usually code for Methionine,
despite what the codon will code for when it
is internal to a protein. This qualifier
sets all such START codons to code for
Methionine by default.
-circular boolean [N] Is the sequence circular
-[no]reverse boolean [Y] Set this to be false if you do not wish
to find ORFs in the reverse complement of
the sequence.
-flanking integer [100] If you have chosen one of the options
of the type of sequence to find that gives
the flanking sequence around a STOP or START
codon, this allows you to set the number of
nucleotides either side of that codon to
output. If the region of flanking
nucleotides crosses the start or end of the
sequence, no output is given for this codon.
(Any integer value)
-sbegin1 integer Start of each sequence to be used
-send1 integer End of each sequence to be used
-sreverse1 boolean Reverse (if DNA)
-snucleotide1 boolean Sequence is nucleotide
-sprotein1 boolean Sequence is protein
-slower1 boolean Make lower case
-supper1 boolean Make upper case
-scircular1 boolean Sequence is circular
Output format"f":
>[Contig]_[ORF.No]_[Start]_[End]_[Strand]\n[Sequence] Manual"t":
[Contig]_[ORF.No]\t[Start]\t[End]\t[Strand]\t[Sequence]
Example usage:
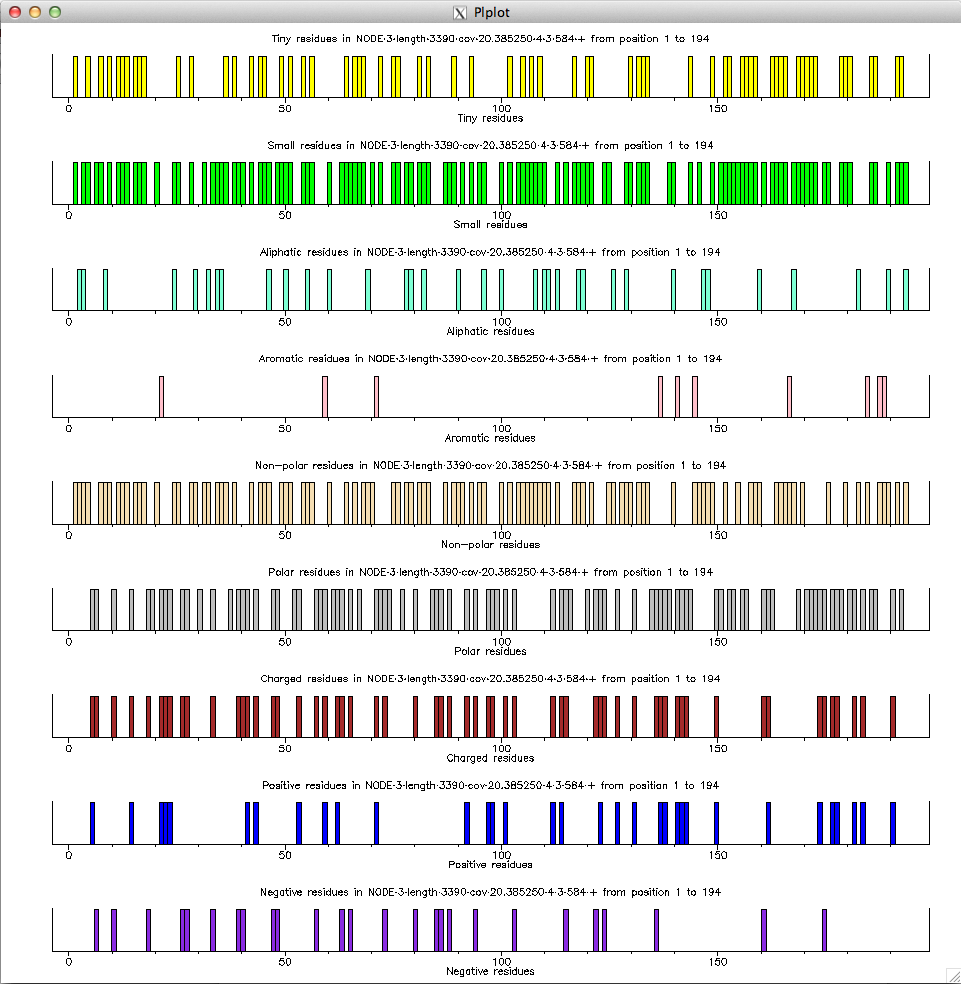
$ echo "NODE_3_length_3390_cov_20.385250" | FASTAgrep --buffer-size=100000000 contigs.fa | ./getorf.sh "f" "-minsize=30 -maxsize=10000" | head -10 >NODE_3_length_3390_cov_20.385250_1_1_234_+ AGSLPATASVKPPPGPVVSSRTGASPRRRCSRSTSSGRQTTAVAPESRTAWRSGASGNSTFSGTPTPPACHTPSRPGR >NODE_3_length_3390_cov_20.385250_2_56_343_+ AAAPARRRGGVAAARRRRAGRRPRSRRSRGRRGAAGRRGTARSAGRRRHRPATHRAGRAGSRGCWAGRTRRAPPVRARAPTGARRTGPRCRRTART >NODE_3_length_3390_cov_20.385250_3_238_504_+ SRVLGRKNPTRSPGASPCADRCAANRAPVSAYCAYVTRVSSRPSATRSAWSDALRRKSIAMFIDDPSLCSAGTPRTRRPRPWTAARRPG >NODE_3_length_3390_cov_20.385250_4_3_584_+ GLVARDGLGEAAARAGGEQPHRRVAEEALQPLDVVGQADDRGRAGVEDGVAQRGVGEQHVQRDADATGLPHTEQAGQVVEGVGQEEPDALPRCEPVRRQVRGEPGPGVGVLRVRDQGVVTAERDPLRMVGRAAAQEHRNVHRRSFPLLCRNASNPSTAALDRSAAAWVTASSSRDPRRSASRLRFTSFFVRATV >NODE_3_length_3390_cov_20.385250_5_588_761_+ SGIAASSWAFATAVASAASASGSTTFAMPSSSARSAPISGASSISSRARCIPTIRGSR $ echo "NODE_3_length_3390_cov_20.385250" | FASTAgrep --buffer-size=100000000 contigs.fa | ./getorf.sh "t" "-minsize=30 -maxsize=10000" | head -10 NODE_3_length_3390_cov_20.385250_1 1 234 + AGSLPATASVKPPPGPVVSSRTGASPRRRCSRSTSSGRQTTAVAPESRTAWRSGASGNSTFSGTPTPPACHTPSRPGR NODE_3_length_3390_cov_20.385250_2 56 343 + AAAPARRRGGVAAARRRRAGRRPRSRRSRGRRGAAGRRGTARSAGRRRHRPATHRAGRAGSRGCWAGRTRRAPPVRARAPTGARRTGPRCRRTART NODE_3_length_3390_cov_20.385250_3 238 504 + SRVLGRKNPTRSPGASPCADRCAANRAPVSAYCAYVTRVSSRPSATRSAWSDALRRKSIAMFIDDPSLCSAGTPRTRRPRPWTAARRPG NODE_3_length_3390_cov_20.385250_4 3 584 + GLVARDGLGEAAARAGGEQPHRRVAEEALQPLDVVGQADDRGRAGVEDGVAQRGVGEQHVQRDADATGLPHTEQAGQVVEGVGQEEPDALPRCEPVRRQVRGEPGPGVGVLRVRDQGVVTAERDPLRMVGRAAAQEHRNVHRRSFPLLCRNASNPSTAALDRSAAAWVTASSSRDPRRSASRLRFTSFFVRATV NODE_3_length_3390_cov_20.385250_5 588 761 + SGIAASSWAFATAVASAASASGSTTFAMPSSSARSAPISGASSISSRARCIPTIRGSR NODE_3_length_3390_cov_20.385250_6 508 795 + RPPAPGTPAGRRRGCGSPASSSGRPCSRASRPARGPSPPPSRARRPRRARRRSRCPAPAPARHRSRGPAASAPVPGASRRSAAAGKPRRRRGRCPA NODE_3_length_3390_cov_20.385250_7 799 831 + RTPRRCGRRRT NODE_3_length_3390_cov_20.385250_8 765 875 + APATSGTMPRLTNTSQMRASADMITRSHTRARLIPPP NODE_3_length_3390_cov_20.385250_9 879 944 + AMPDTAATTGSRDSQISTTSRW NODE_3_length_3390_cov_20.385250_10 865 1047 + SRRRRRCPTPPRPRAAATPRSRRRAGGRPASSSGARRRRHRRRCRGCCRRPRPRRSARRSRYou can then use pepinfo to draw amino-acid properties for
NODE_3_length_3390_cov_20.385250_4_3_584_+ by saving the sequence in a file test.faa and calling it as follows:
$ pepinfo test.faa -auto
Last Updated by Dr Umer Zeeshan Ijaz on 20/04/2014.