Metaproteomics Data Analysis Workflow
by Umer Zeeshan Ijaz, Florence Abram, Aoife Joyce, and Chris Quince
This tutorial gives a step-by-step description of the metaproteomics data analysis workflow. We start off with ProteinPilot, a software tool for protein identification and quantitation that uses a hybrid sequence tag and database search approach using feature probabilities to identify hundreds of sequence variant in a single step for the data generated from mass spectrometers. The software uses peptide results, condenses it down to the most defensible set of detected proteins with ambiguity and reports their accession numbers (Uniprot) along with quality statistics on peptide and protein identification. The final results can be exported as an excel file:
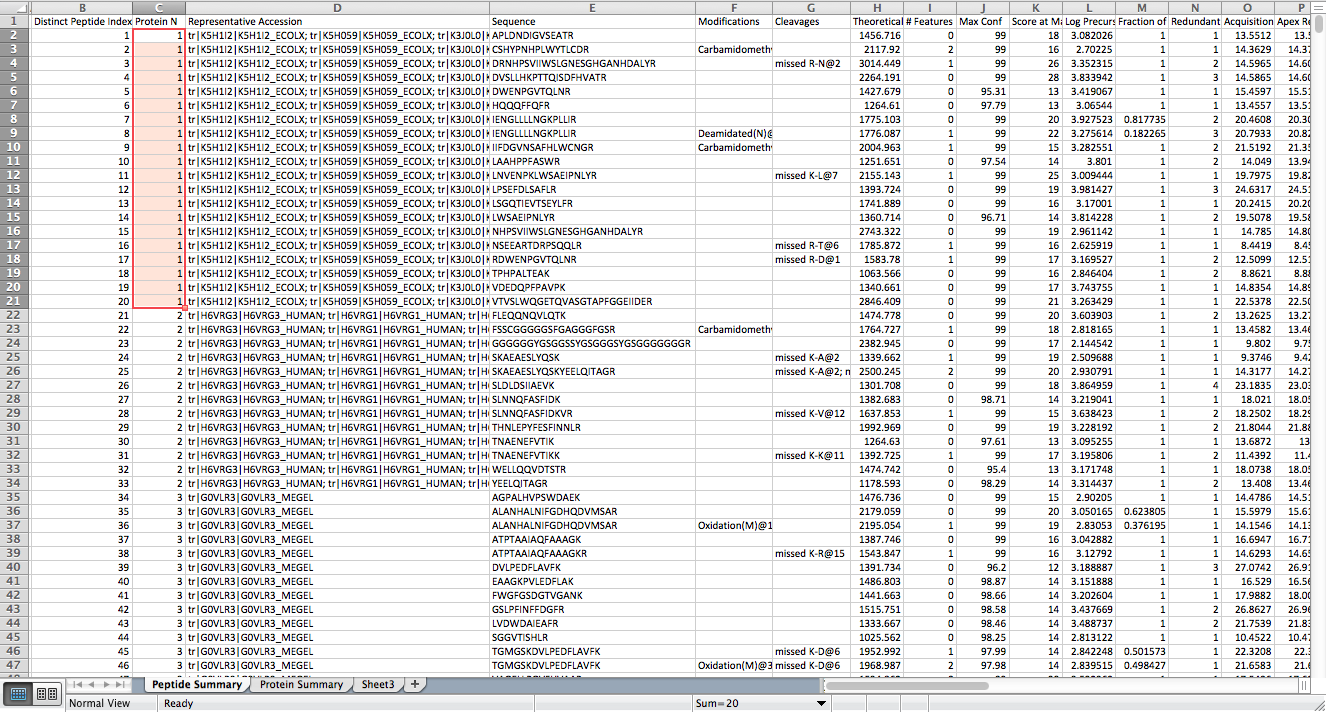
In the above figure, assignment of peptide sequences to different proteins are shown in "Protein N" column in the first sheet. For example, the first protein contains 20 such sequences. The "Representative Accession" lists all the ambigious protein assignments identifiable Uniprot IDs.
We have written the processPROT.py script that can read the Excel files generated from ProteinPilot, download the protein sequences and associated metadata from Swiss-Prot using ExPASy server, and filter out any proteins for which there is not enough supporting evidence (less than 2 peptides sequences assigned).
[uzi@quince-srv2 ~/Metaproteomics]$ ./processPROT.py -i Sample_1_a.xls -i Sample_1_b.xls -i Sample_1_c.xls > Sample_1.tsv
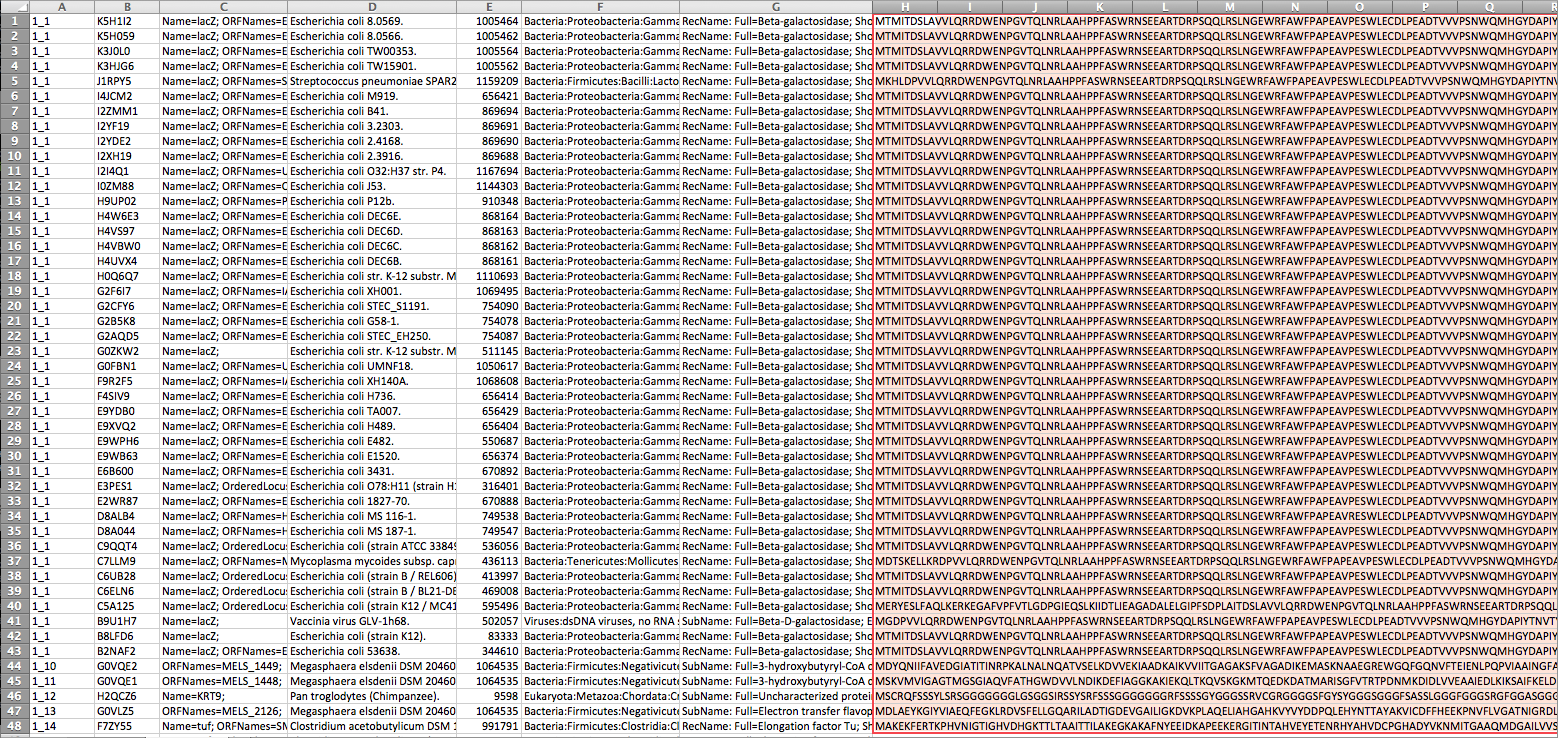
You can use -i switch to provide as many Excel files as you have for a given sample, with the tab-delimited output file listing proteins with unique numeric identifiers with the format [Sample ID]_[Protein ID] as shown in the first column in the following figure. We output a 5-tuple record (ID, Uniprot Identifier, Name, Assignment to Organism, Taxonomy ID, Classification Path of the Organism, Protein Sequence). Columns with missing data have __NA__ value in them.
Since the last column contains the protein sequences, we can use the following one-liner to extract the unique protein sequences in the form of a FASTA file. Since, we are only interested in microbial communities, we also remove any "Eukaryota" entries from the table:
[uzi@quince-srv2 ~/Metaproteomics]$ grep -vi "Eukaryota" Sample_1.tsv | awk -F"\t" '$8!="__NA__"{print $8}' | sort | uniq | awk '{print ">PROT"++i"\n"$0}' > Sample_1.fasta
[uzi@quince-srv2 ~/Metaproteomics]$ head Sample_1.fasta
>PROT1
AAPAAAAPAAAPAAQAAAPAAAGGDDAILSPAARKLAEEAGIDPNSIAGTGKGGRVTKEDVVAAVEAKKNAPAALAKPAAPAAEAPIFAAGDRVEKRVPMTRLRAKVAERLVEAQSAMAMLTTFNEVNMKPIMDLRSKYKDLFEKKHNGVRLGFMSFFVKAATEALKRFPGVNASIDGNDIVYHGYQDIGVAVSSDRGLVVPVLRNAEFMSLAEIEGGIANFGKKAKEGKLTIEDMTGGTFTISNGGVFGSLLSTPIVNPPQTAILGMHKIQERPMAVNGQVVILPMMYLALSYDHRLIDGKEAVSFLVAIKDLLEDPARLLLDV
>PROT2
AAPAAAGGDDAILSPAARKLAEEAGIDPNSIAGTGKGGRVTKEDVVAAVEAKKNAPAAPAKPAAPAAEAPIFAAGDRVEKRVPMTRLRAKVAERLVEAQSAMAMLTTFNEVNMKPIMDLRSKYKDLFEKKHNGVRLGFMSFFVKAATEALKRFPGVNASIDGNDIVYHGYQDIGVAVSSDRGLVVPVLRNAEFMSLAEIEGGIANFGKKAKEGKLTIEDMTGGTFTISNGGVFGSLLSTPIVNPPQTAILGMHKIQERPMAVNGQVVILPMMYLALSYDHRLIDGKEAVSFLVAIKDLLEDPARLLLDV
>PROT3
AKSKFERNKPHVNIGTIGHVDHGKTSLTAAITKYFGEFKAYDQIDAAPEEKARGITISTAHVEYETPNRHYAHVDCPGHADYVKNMITGAAQMDGAILVVSAADGPMPQTREHILLARQVGVPAIVVFLNKVDQVDDAELLELVELEVRELLSSYEFPGDDIPIVKGSALAALEDSDKKIGEDAIRELMAAVDAYIPTPERPIDQPFLMPIEDVFSISGRGTVVTGRVERGIVKVGEEIEIVGIRPTTKTTCTGVEMFRKLLDQGQAGDNIGALLRGVDRNGVERGQILCKPGSVKPHRKFKAEAYILTKEEGGRHTPFFTNYRPQFYFRTTDVTGIVTLPEGTEMVMPGDNVTVDVELIVPIAMEEKLRFAIREGGRTVGAGIVASIVE
>PROT4
APAAAAPAAAPAAQAAAPAAAGGDDAILSPAARKLAEEAGIDPNSIAGTGKGGRVTKEDVVAAVEAKKNAPAAPAKPAAPAAEAPIFAAGDRVEKRVPMTRLRAKVAERLVEAQSAMAMLTTFNEVNMKPIMDLRSKYKDLFEKKHNGVRLGFMSFFVKAATEALKRFPGVNASIDGNDIVYHGYQDIGVAVSSDRGLVVPVLRNAEFMSLAEIEGGIANFGKKAKEGKLTIEDMTGGTFTISNGGVFGSLLSTPIVNPPQTAILGMHKIQERPMAVNGQVVILPMMYLALSYDHRLIDGKEAVSFLVAIKDLLEDPARLLLDV
>PROT5
ATLLAQAIIREGMKNVTAGANPMVVRKGIQDAVDAAVEALHKNSKKVTCSEDIARVATISAGDETVGKLIAEAMEKVTSDGVITVEESKTSETYSEVVEGMMFDRGYISPYMATDTEKMEAIMDDAYLLITDKKISNIQEILPLLEKIVQSGKKLVIIAEDVEGEALTTLILNKLRGTFNCAAV
Next, we use RAPsearch to search our protein sequences against the KEGG database and get a tab-delimited blast output file (identified by *.out.m8 ending). To speed up RAPsearch, you can specify the number of cores to run on using -z switch.
[uzi@quince-srv2 ~/Metaproteomics]$ rapsearch -d /home/opt/keggs_database/kegg_db -z 2 -q Sample_1.fasta -o Sample_1.out
[uzi@quince-srv2 ~/Metaproteomics]$ head *.out.m8
# RAPSearch
# Job submitted: Thu Jul 3 17:57:51 2014
# Query : Sample_1.fasta
# Subject : /home/opt/keggs_database/kegg_db
# Fields: Query Subject identity aln-len mismatch gap-openings q.start q.end s.start s.end log(e-value) bit-score
PROT1 psa:PST_1876 89.2308 325 35 0 1 325 84 408 -156.9 557.37
PROT1 pmy:Pmen_2502 84.7095 327 48 1 1 325 84 410 -149.71 533.49
PROT1 psb:Psyr_2010 78.7692 325 67 1 1 325 89 411 -137.65 493.43
PROT1 hch:HCH_04744 73.2143 336 74 4 2 325 80 411 -130.46 469.54
PROT1 sde:Sde_2105 71.6049 324 89 2 5 325 80 403 -124.55 449.9
We are only interested in finding the general function of microbial community. This is done by identifying enzymes in our dataset. For this purpose, we use a REST-style KEGG API to link KEGG entries (2nd column in the output file) to EC numbers:
[uzi@quince-srv2 ~/Metaproteomics]$ awk -F"\t" '!/#/{print $1","$2}' Sample_1.out.m8 | while IFS=$"," read -r -a mA; do echo -e "${mA[0]}\t$(curl -s http://rest.kegg.jp/link/ec/${mA[1]})"; done | awk '{print $1"\t"$3}' > ec_assignments.txt
[uzi@quince-srv2 ~/Metaproteomics]$ head ec_assignments.txt
PROT1 ec:2.3.1.61
PROT1 ec:2.3.1.61
PROT1 ec:2.3.1.61
PROT1 ec:2.3.1.61
PROT1 ec:2.3.1.61
PROT1 ec:2.3.1.61
PROT1 ec:2.3.1.61
PROT1 ec:2.3.1.61
PROT1 ec:2.3.1.61
PROT1 ec:2.3.1.61
RAPsearch reports multiple matches for a single protein sequence. We assign the most abundant EC number to each protein sequence:
[uzi@quince-srv2 ~/Metaproteomics]$ sort ec_assignments.txt | uniq -c | grep -i "ec:" | awk '{gsub("^ec:","",$3);if(ec[$2]){if($1>=c[$2]){ec[$2]=$3;c[$2]=$1}} else {ec[$2]=$3;c[$2]=$1}} END{for (i in ec) print i"\t"ec[i]}' > Sample_1.ec
[uzi@quince-srv2 ~/Metaproteomics]$ tail Sample_1.ec
PROT1 2.3.1.61
PROT78 4.2.1.11
PROT2 2.3.1.61
PROT136 1.1.1.86
PROT137 1.12.7.2
PROT4 2.3.1.61
PROT138 2.7.9.1
PROT139 3.2.1.23
PROT95 1.1.1.86
PROT9 3.6.3.14
We next construct parsimonious pathways using MinPath as it does not attempt to reconstruct entire pathways from a given set of protein sequences identified in a protemics experiment, but determines the minimal set of biological pathways that must exist in the biological system to explain the input protein sequences. It takes a set of reference pathways and a set of proteins that can be mapped to one or more pathways, and finds the minimum number of pathways that can explain all the protein functions.
We will using MinPath
To use MinPath, you need to setup environmental variables in your ~/.bashrc file after downloading the software
# Required for minpath
export PATH=/home/opt/MinPath:$PATH
export MinPath=/home/opt/MinPath
MinPath comes with a mapping file ec2path that lists the reference pathways from MetaCyc and all the EC numbers associated with them. MetaCyc serves as an encyclopedia of metabolism containing more than 2151 patways from more than 2500 different organisms.
[uzi@quince-srv2 /home/opt/MinPath/data]$ head -50 ec2path
#Metacyc pathway and ec mapping file
#Pathway EC
PWY0-1479 3.1.26.12
PWY0-1479 2.7.7.56
PWY0-1479 3.1.13.5
PWY0-1479 3.1.26.5
PWY0-1479 3.1.13.5
PWY0-1479 2.7.7.56
PWY0-1479 3.1.26.5
PWY0-1479 3.1.13.1
PWY0-1479 3.1.13.1
PWY0-1479 3.1.26.12
PWY-6146 4.1.1.31
PWY-6146 4.2.1.1
PWY-6146 6.4.1.1
PWY-6146 4.1.1.31
PWY-6146 2.7.1.40
PWY-6146 4.2.1.11
PWY-6146 5.4.2.1
PWY-6146 1.2.7.6
PWY-6146 5.3.1.1
PWY-6146 4.1.2.13
PWY-6146 2.7.1.146
PWY-6146 5.3.1.9
PWY-6146 1.2.7.1
PWY-6146 1.2.7.4
PWY-6146 2.1.1.-
PWY-6146 1.5.1.5
PWY-6146 3.5.4.9
PWY-6146 6.3.4.3
PWY-6146 1.2.1.43
PWY-6146 1.1.1.37
PWY-6146 1.3.99.1
PWY-6146 1.2.7.3
PWY-6146 6.2.1.5
PWY-6146 4.2.1.2
PWY-6146 6.4.1.1
PWY-6146 1.2.7.1
PWY-6146 2.6.1.1
PWY-6146 2.6.1.2
PWY-6146 1.1.1.261
PWY-6146 2.5.1.41
PWY-6146 2.5.1.42
PWY-6146 2.7.7.67
PWY-6146 2.5.1.29
PWY-6146 5.3.3.2
PWY-6146 2.5.1.10
PWY-6146 2.5.1.1
PWY-6146 5.3.3.2
PWY-6146 4.1.1.33
To construct pathways from the generated EC file using MetaCyc pathways (using default mapping file in MinPath), run it as follows:
[uzi@quince-srv2 ~/Metaproteomics]$ MinPath1.2.py -any Sample_1.ec -map ec2path -report Sample_1.ec.report -details Sample_1.ec.details
[uzi@quince-srv2 ~/Metaproteomics]$ head Sample_1.ec.report
path 2 any n/a naive 1 minpath 0 fam0 39 fam-found 2 name PWY-6146
path 4 any n/a naive 1 minpath 1 fam0 8 fam-found 1 name BRANCHED-CHAIN-AA-SYN-PWY
path 11 any n/a naive 1 minpath 1 fam0 9 fam-found 1 name PWY-821
path 44 any n/a naive 1 minpath 1 fam0 1 fam-found 1 name GLUTSYNIII-PWY
path 46 any n/a naive 1 minpath 0 fam0 6 fam-found 1 name PWY-5505
path 49 any n/a naive 1 minpath 1 fam0 8 fam-found 1 name PWY-6549
path 53 any n/a naive 1 minpath 0 fam0 13 fam-found 1 name THREOCAT-PWY
path 55 any n/a naive 1 minpath 0 fam0 5 fam-found 1 name ILEUSYN-PWY
path 56 any n/a naive 1 minpath 0 fam0 11 fam-found 1 name PWY-3001
path 57 any n/a naive 1 minpath 0 fam0 6 fam-found 1 name PWY-5101
The last column reports the pathways, which you can search on MetaCyc website to get the detailed information. For example, here is the record for BRANCHED-CHAIN-AA-SYN-PWY on the website:

However, we are interested in KEGG pathways so that we can draw metabolic networks using iPath. We, therefore, generate a new mapping file using the REST-style KEGG API:
[uzi@quince-srv2 /home/opt/MinPath/data]$ for i in $(curl -S http://rest.kegg.jp/list/pathway | awk -F"\t" '{print $1}'); do curl -S http://rest.kegg.jp/link/ec/$i; done > ec2kegg
[uzi@quince-srv2 /home/opt/MinPath/data]$ awk -F"\t" '!/^$/{gsub("^path:","",$1);gsub("^ec:","",$2);print $1"\t"$2}' ec2kegg > ec2kegg_final
[uzi@quince-srv2 /home/opt/MinPath/data]$ chmod +x ec2kegg_final
[uzi@quince-srv2 /home/opt/MinPath/data]$ head -50 ec2kegg_final
map00010 1.1.1.1
map00010 1.1.1.2
map00010 1.1.1.27
map00010 1.1.2.7
map00010 1.1.2.8
map00010 1.2.1.12
map00010 1.2.1.3
map00010 1.2.1.5
map00010 1.2.1.59
map00010 1.2.1.9
map00010 1.2.4.1
map00010 1.2.7.1
map00010 1.2.7.6
map00010 1.8.1.4
map00010 2.3.1.12
map00010 2.7.1.1
map00010 2.7.1.11
map00010 2.7.1.146
map00010 2.7.1.147
map00010 2.7.1.2
map00010 2.7.1.40
map00010 2.7.1.41
map00010 2.7.1.63
map00010 2.7.1.69
map00010 2.7.2.3
map00010 3.1.3.10
map00010 3.1.3.11
map00010 3.1.3.13
map00010 3.1.3.9
map00010 3.2.1.86
map00010 4.1.1.1
map00010 4.1.1.32
map00010 4.1.1.49
map00010 4.1.2.13
map00010 4.2.1.11
map00010 5.1.3.15
map00010 5.1.3.3
map00010 5.3.1.1
map00010 5.3.1.9
map00010 5.4.2.11
map00010 5.4.2.12
map00010 5.4.2.2
map00010 5.4.2.4
map00010 6.2.1.1
map00010 6.2.1.13
map00020 1.1.1.286
map00020 1.1.1.37
map00020 1.1.1.41
map00020 1.1.1.42
map00020 1.1.5.4
Now run MinPath1.2.py with the new mapping file using -map ec2kegg_final in the command:
[uzi@quince-srv2 ~/Metaproteomics]$ MinPath1.2.py -any Sample_1.ec -map ec2kegg_final -report Sample_1.ec.report.kegg -details Sample_1.ec.details.kegg
[uzi@quince-srv2 ~/Metaproteomics]$ head Sample_1.ec.report.kegg
path 1 any n/a naive 1 minpath 0 fam0 45 fam-found 3 name map00010
path 2 any n/a naive 1 minpath 0 fam0 24 fam-found 1 name map00020
path 3 any n/a naive 1 minpath 0 fam0 50 fam-found 1 name map00030
path 4 any n/a naive 1 minpath 0 fam0 63 fam-found 1 name map00040
path 5 any n/a naive 1 minpath 0 fam0 70 fam-found 2 name map00051
path 6 any n/a naive 1 minpath 0 fam0 46 fam-found 1 name map00052
path 9 any n/a naive 1 minpath 0 fam0 13 fam-found 1 name map00062
path 10 any n/a naive 1 minpath 0 fam0 30 fam-found 2 name map00071
path 18 any n/a naive 1 minpath 0 fam0 11 fam-found 2 name map00190
path 19 any n/a naive 1 minpath 0 fam0 3 fam-found 1 name map00195
To get the description of the reported KEGG pathways, use the following one-liner:
[uzi@quince-srv2 ~/Metaproteomics]$ for i in $(awk '{print $14}' Sample_1.ec.report.kegg); do curl -S http://rest.kegg.jp/find/pathway/$i; done
path:map00010 Glycolysis / Gluconeogenesis
path:map00020 Citrate cycle (TCA cycle)
path:map00030 Pentose phosphate pathway
path:map00040 Pentose and glucuronate interconversions
path:map00051 Fructose and mannose metabolism
path:map00052 Galactose metabolism
path:map00062 Fatty acid elongation
path:map00071 Fatty acid degradation
path:map00190 Oxidative phosphorylation
path:map00195 Photosynthesis
path:map00230 Purine metabolism
path:map00240 Pyrimidine metabolism
path:map00250 Alanine, aspartate and glutamate metabolism
path:map00280 Valine, leucine and isoleucine degradation
path:map00281 Geraniol degradation
path:map00290 Valine, leucine and isoleucine biosynthesis
path:map00310 Lysine degradation
path:map00330 Arginine and proline metabolism
path:map00360 Phenylalanine metabolism
path:map00362 Benzoate degradation
path:map00380 Tryptophan metabolism
path:map00410 beta-Alanine metabolism
path:map00511 Other glycan degradation
path:map00531 Glycosaminoglycan degradation
path:map00592 alpha-Linolenic acid metabolism
path:map00600 Sphingolipid metabolism
path:map00604 Glycosphingolipid biosynthesis - ganglio series
path:map00620 Pyruvate metabolism
path:map00627 Aminobenzoate degradation
path:map00630 Glyoxylate and dicarboxylate metabolism
path:map00640 Propanoate metabolism
path:map00650 Butanoate metabolism
path:map00680 Methane metabolism
path:map00710 Carbon fixation in photosynthetic organisms
path:map00720 Carbon fixation pathways in prokaryotes
path:map00730 Thiamine metabolism
path:map00770 Pantothenate and CoA biosynthesis
path:map00903 Limonene and pinene degradation
path:map00910 Nitrogen metabolism
path:map00920 Sulfur metabolism
path:map00930 Caprolactam degradation
path:map01040 Biosynthesis of unsaturated fatty acids
path:map01100 Metabolic pathways
path:map01110 Biosynthesis of secondary metabolites
path:map01120 Microbial metabolism in diverse environments
We want to use interactive Pathways Explorer (iPath), which is a web-based tool for the visualization, analysis and customization of the various pathways maps. The metabolic pathways are constructed using 146 KEGG pathways, and give an overview of the complete metabolism in biological systems.
We first extract the pathways in the format to be used with iPath and give each of them red color:
[uzi@quince-srv2 ~/Metaproteomics]$ awk '{gsub("^map","",$14);print $14" #ff0000"}' Sample_1.ec.report.kegg > Sample_1.ec.report.kegg.ipath
Now go to http://pathways.embl.de/index.html and click on iPath v2 interface to load the applet:

Next click on the "Customize" button to load the "Customization and data mapping" tab, and upload Sample_1.ec.report.kegg.ipath by clicking on "Select file" button. This will populate the "Element selection" text area. You can manually assign different colors to your pathways here. Finally, click on "Submit data and customize maps" button to generate your metabolic network.

Once generated, you can save the metabolic network as a PDF document to your local computer. The connections in red are the ones present in your sample:

You can then repeat the same analysis on your other samples.
Last Updated by Dr Umer Zeeshan Ijaz on 05/07/2014.